Maligner peripherer Nervenscheidentumor des Unterkiefers
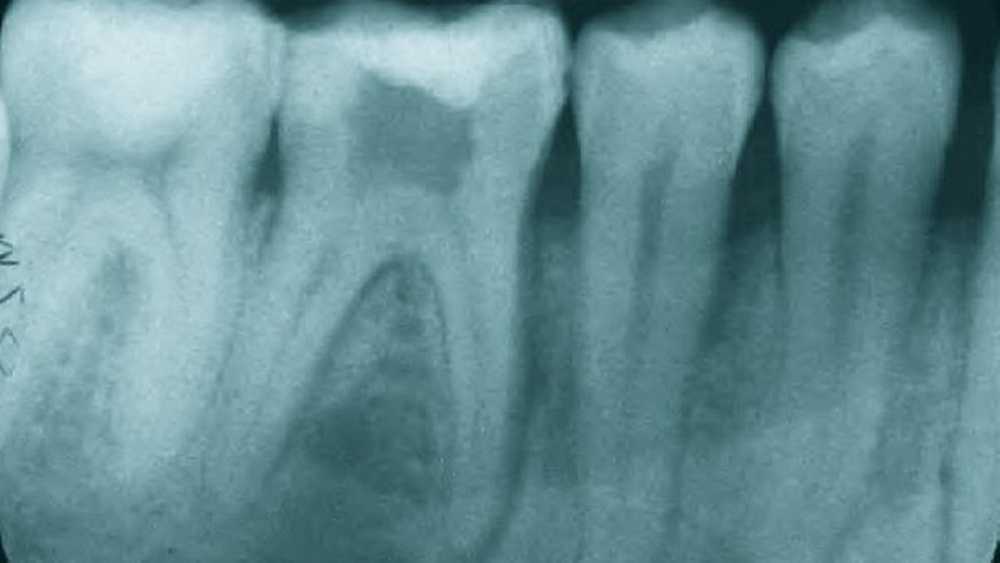
- Abbildung 1: Zahnfilm 44-47: Apikale Aufhellung, erweiterter PA-Spalt und interradikuläre Osteolyse an Zahn 46



Circa zehn Prozent aller Weichteilsarkome sind MPNST. 50 Prozent der Tumoren werden als de novo Synthese und 50 Prozent bei Patienten mit Vorliegen einer Neurofibromatose Typ-1 oder bei Zustand nach Strahlentherapie gefunden. Der Altersmedian liegt bei 35 Jahren, Männer sind viermal häufiger betroffen als Frauen [1]. Maligne periphere Nervenscheidentumoren sind meist schmerzhaft und fest im umgebenden Gewebe verankert [2]. Ihr schnelles Wachstum, die hohe Fernmetastasierungsrate (Skelett, lymphatisches System, Lunge, Leber) von 40 bis 80 Prozent und die hohe Lokalrezidivrate nach radikaler Exzision von circa 50 Prozent bedeuten eine schlechte Prognose: Die Fünf-Jahres-Überlebensrate liegt bei 53 Prozent, die Zehn-Jahres-Überlebensrate bei 34 Prozent [6,7]. Die Lokalisation dieses Tumors im Unterkiefer ist extrem selten.
Fallbericht
Ein 24-jähriger Patient stellte sich mit zunehmenden Beschwerden im Bereich des rechten Unterkiefers bei seinem Zahnarzt vor. Nach einer in Bezug auf die Beschwerdesymptomatik erfolglosen Füllungs-, endodontischen und schließlich Extraktionstherapie des vermeintlich schuldigen Zahnes 46 (Abbildung 1) wurde der Patient in die Abteilung für Mund-, Kiefer- und Gesichtschirurgie des Universitätsklinikums Freiburg überwiesen.
Der Patient stellte sich in gutem Allgemein- und Ernährungszustand vor. Kopf und Hals sowie Haut und sichtbare Schleimhäute waren bis auf den Bereich des rechten Unterkiefers unauffällig. In diesem Gebiet berichtete der Patient über persistierende Schmerzen. In der Familienanamnese waren bislang keine onkologischen Erkrankungen bekannt.
Der Lokalbefund des rechten Unterkiefers zeigte enoral eine eitrige, gerötete Wunde bei Zustand nach Zahnextraktion mit sublingualer Schleimhautinduration (Abbildung 2). Neurologisch auffällig war eine nach Bericht des Patienten zunehmende Hypästhesie im Ausbreitungsgebiet des N. mentalis rechts. Sonographisch waren keine peripheren Lymphknotenvergrößerungen feststellbar.
Die präoperative Panoramaschichtaufnahme zeigte zwei ausgedehnte osteolytische Areale in Regio 33 und 46. In Regio 35 bis 48 zeigte sich ein disseminiert osteolytisch veränderter Knochen mit multiplen periapikalen Aufhellungen (Abbildung 3).
Die Biopsie zeigte histopathologisch einen spindeligen, mesenchymalen Tumor, der einem hoch malignen Sarkom entsprach. Die Typisierung gestaltete sich kompliziert, da der Tumor initial auch durch immunhistochemische Untersuchungen nicht eindeutig zu klassifizieren war (siehe unten).
Die zum Teil knotig aufgebaute Läsion zeigte Infiltrate eines zellreichen, hoch malignen, spindeligen, gelegentlich faszikelartig wachsenden Tumors mit herdförmig angeordneten, größeren epitheloiden Zellen. Die überwiegend irregulär angeordneten Formationen wiesen Zellen mit polymorphen, atypischen Zellkernen auf, die bis zu 20 atypische Mitosen auf zehn High-Power-Fields (HPF, Hauptgesichtsfeld bei 400- facher Vergrößerung) zeigten. Es imponierten ausgedehnte Koagulationsnekrosen und eine Destruktion des ortsständigen Knochens (Abbildungen 4, 5, 6).
Immunhistochemisch exprimierten die Zellen gleichzeitig epitheliale (Zytokeratin 7 und 19) und mesenchymale (Vimentin, Alpha-Aktin) Marker (Abbildungen 7, 8), sodass man zusammen mit der Morphologie und trotz der ungewöhnlichen, jedoch durchaus möglichen Lokalisation an ein monophasisches spindelzelliges synoviales Sarkom dachte. Die Fluoreszenz-in-situ-Hybridisierung zeigte jedoch keine für diese Entität typische Translokation im Bereich des SYT-Gens (18q11) [3].
Unter Berücksichtigung der klinischen Symptomatik (Schmerzen, Hypästhesie), der histologisch nachweisbaren Affinität zu Nerven und Gefäßen, der Positivität für Desmin sowie der Absenz von Mastzellen wurde schließlich die Diagnose eines malignen peripheren Nervscheidentumors gestellt.
Im Sinne eines Stagings erfolgte vor geplanter präoperativer Chemotherapie neben einem Röntgen-Thorax eine Magnetresonanztomographie (MRT) des Abdomens und des Halses, eine Computertomographie (CT) des Thorax, sowie eine Positronenemissionstomographie (PET) und eine Beckenkammbiopsie. MRT und CT des Schädels zeigten einen ausgedehnten Tumor im rechten Unterkiefer mit Osseodestruktion im Bereich des rechten Kieferwinkels bis in die linke Prämolarenregion. Weichgewebig zeigte sich ein verdrängendes Wachstum mit Infiltration des Mundbodens und des M. masseter, sowie des M. pterygoideus medialis rechts (Abbildungen 9 und 10).
Thoraxübersicht, MR-Abdomen, CT-Thorax sowie eine Beckenkammbiopsie zeigten keinen Anhalt auf Fernmetastasen. Die PET markierte einen deutlich erhöhten Metabolismus des Tracers (Fluordesoxyglukose) im Bereich des rechten Unterkiefers (Abbildung 11).
Die Behandlung des Patienten erfolgte nach Besprechung der Befunde in einer interdisziplinären Tumorkonferenz der Universitätsklinik Freiburg in Zusammenarbeit der Abteilung für Hämatologie und Onkologie, der Mund-, Kiefer- und Gesichtschirurgie, der Klinik für Strahlenheilkunde und dem Institut für Pathologie.
Entsprechend der Cooperativen Weichteilsarkomstudie (CWS 2002-P) für Kinder und Jugendliche, erfolgte durch die Onkologie zunächst die neoadjuvante Chemotherapie analog der Hochrisikogruppe für lokalisierte nicht rhabdomyosarkomartige Sarkome (Non-RMS-Sarkome). Die Chemotherapie wurde gemäß dem VAIA-III-Protokoll mit vier Blöcken I2VAd mit den Medikamenten Ifosfamid, Vincristin, Adriamycin (Block 1, 3, 4, 6) und fünf Blöcken I2VA mit den Medikamenten Ifosfamid, Vincristin und Actinomycin D (Block 2, 5, 7, 8, 9) geplant.
Im Anschluss wurde der Tumor durch eine ausgedehnte Unterkieferteilresektion vom aufsteigenden Unterkieferast rechts bis Regio Zahn 36 im Gesunden entfernt. Der resezierte Unterkieferkörper wurde primär alloplastisch durch eine Rekonstruktionsplatte ersetzt. Das Tumorlager wurde intraoperativ mit 16 Gy bestrahlt (Abbildung 12).
Die abschließende Pathologie des Resektionspräparates zeigte einen im Gesunden entfernten malignen peripheren Nervenscheidentumor (FNCLCC-Klassifikation: Grad 3) mit der Tumorformel (UICC) ypT2b, pN0 (0/4), pMx, pNx, L0, V0, R0. Der Tumor wies ausgeprägte regressive Veränderungen auf. Die histologisch gesicherte Tumorgröße maß im Weichgewebe maximal 4,9 cm und in den Markräumen des Unterkiefers zirka zehn Zentimeter. Postoperativ erfolgten weitere strahlen- und chemotherapeutische Maßnahmen. Drei Monate nach Resektion stellte sich die Situation klinisch und röntgenologisch stabil und ohne Anhalt für ein Rezidiv dar (Abbildungen 13 a+b).
Weitere drei Monate später zeigten sich dorsal der rechten Kieferhöhle eine Kontrastmittel aufnehmende Raumforderung mit Verdacht auf ein Lokalrezidiv (Abbildung 14) sowie multiple pulmonale und pleurale Raumforderungen, die bioptisch als Metastasen des bekannten Tumors diagnostiziert wurden (Abbildungen 15 a+b).
Diskussion
Die meisten von Nervenscheiden ausgehenden Tumoren sind benigner Natur (Schwannome, Neurofibrome), die malignen Formen (MPNST, Fibrosarkome) sind insgesamt äußerst selten. Befallen werden große und mittelgroße Nerven meist im Oberschenkel- und Gesäßbereich. Ein Vorkommen im Kopf- und Halsbereich stellt somit eine ausgesprochene Rarität dar und bedingt das Fehlen klinischer Studien und Therapiekonzepte.
Da diese Tumoren erst ab einer Größe zwischen fünf und zehn Zentimetern symptomatisch werden, wird die Erstdiagnose wie auch beim vorliegenden Fall oftmals erst in einem bereits fortgeschrittenen Stadium gestellt. Der Therapiebeginn kann zudem durch eine langwierige und schwierige histologische Diagnosestellung verzögert werden.
Die derzeit für MPNST gültige Therapieempfehlung besteht in der radikalen Exzision des Tumors mit einem Sicherheitsabstand von fünf Zentimetern und adjuvanter Strahlentherapie, wobei keine signifikanten Unterschiede der Erfolgsrate hinsichtlich Brachytherapie, externer Bestrahlung oder Elektronenbestrahlung ausgemacht werden konnten [4, 5, 7]. Dem geforderten Sicherheitsabstand kann im Kopf- und Halsbereich aus vitalen Gründen und anatomischen Gegebenheiten nur eingeschränkt Folge geleistet werden. Im Hinblick auf eine adjuvante Chemotherapie gibt es aktuell keine aussagekräftigen Empfehlungen. Die vorliegenden Studien sind aufgrund unterschiedlicher Chemotherapieprotokolle und uneinheitlicher Histologien nicht vergleichbar. Aufgrund des jungen Alters des Patienten wurde analog der Cooperativen Weichteilsarkomstudie (CWS 2002-P) für Kinder und Jugendliche eine neoadjuvante und adjuvante Chemotherapie durchgeführt, da man sich eine Verbesserung des rezidivfreien Überlebens und Reduktion der Radikalität von Lokalmaßnahmen erhoffte.
Die Tumorerkrankung des Patienten zeigte, dass die Diagnose eines Weichteilsarkoms erschwert ist und spezielle histopathologische Untersuchungsverfahren zur Diagnostik benötigt werden. Im vorliegenden Patientenfall wurde die Diagnose mit Unterstützung des Knochentumor-Referenzzentrums des Kantonspitals Basel (Prof. Dr. Gernot Jundt) bestätigt. Ebenfalls sollte aufgrund der Seltenheit von Sarkomen die Therapie in einem Zentrum durchgeführt werden, welches in Behandlungen dieser Art erfahren ist und die enge Zusammenarbeit zwischen den einzelnen Fachrichtungen gewährleistet.
Als Empfehlung für die zahnärztliche Praxis sollen die Vorzüge von Panoramaschichtaufnahmen im Hinblick auf Fokussuche und Zufallsbefunde in der Routineuntersuchung genannt sein. Neurologische Auffälligkeiten im Kopf- und Halsbereich (Hypästhesien, Paresen, Neuralgien und mehr) ebenso wie unklare Schmerzen oder Raumforderungen bedürfen der genauen Abklärung, um etwaige Neoplasien einer frühzeitigen Therapie zuzuführen.
Dr. Bastian SchmiedProf. Dr. Dr. Ralf SchönUniversitätsklinikum FreiburgAbt. Klinik für MKiG-ChirurgieHugstetter Str. 5579106 Freiburg i. Brsg.bastian.schmied@uniklinik-freiburg.de
Dr. Jürgen HeinzUniversitätsklinikum FreiburgKlinik für Hämatologie und OnkologieAbt. Innere Medizin IHugstetter Str. 5579106 Freiburg i. Brsg.
Dr. Marzenna Orlowska-VolkUniversitätsklinikum FreiburgAbteilung Allgemeine Pathologie undPathologische AnatomieBreisacher Str. 115a79106 Freiburg


{kind=link}
{kind=link}