Raumforderung am Kinn: Histologie zeigt seltenen Befund
Eine 19-jährige Frau stellte sich nach Überweisung in der Poliklinik der Mund-, Kiefer- und Gesichtschirurgie der Universitätsmedizin Mainz vor. Aufgrund einer Raumforderung im Bereich des Kinns hatte sie zuvor einen niedergelassenen MKG-Chirurgen aufgesucht. Dort war durch den Kollegen zunächst die klinische Verdachtsdiagnose eines Lipoms gestellt worden.
Es folgte die Überweisung an unsere Poliklinik mit der Bitte zur weiteren Diagnostik und Therapieübernahme. Hier präsentierte sich die Patientin in einem sehr guten Allgemein- und Ernährungszustand. Anamnestisch konnten keine Grunderkrankungen oder zurückliegenden Operationen eruiert werden. Auch ästhetische Eingriffe wurden in der Krankengeschichte nicht gefunden.
Die Patientin berichtete, dass die Raumforderung mit beginnender Adoleszenz im Alter von 14 Jahren das erste Mal bewusst wahrgenommen wurde. In Anbetracht der geringen Größe wurde dem Befund vom betreuenden Pädiater aber zunächst kein Krankheitswert zugemessen. Die Patientin erhielt im Rahmen von Arztbesuchen regelmäßige klinische Kontrollen.
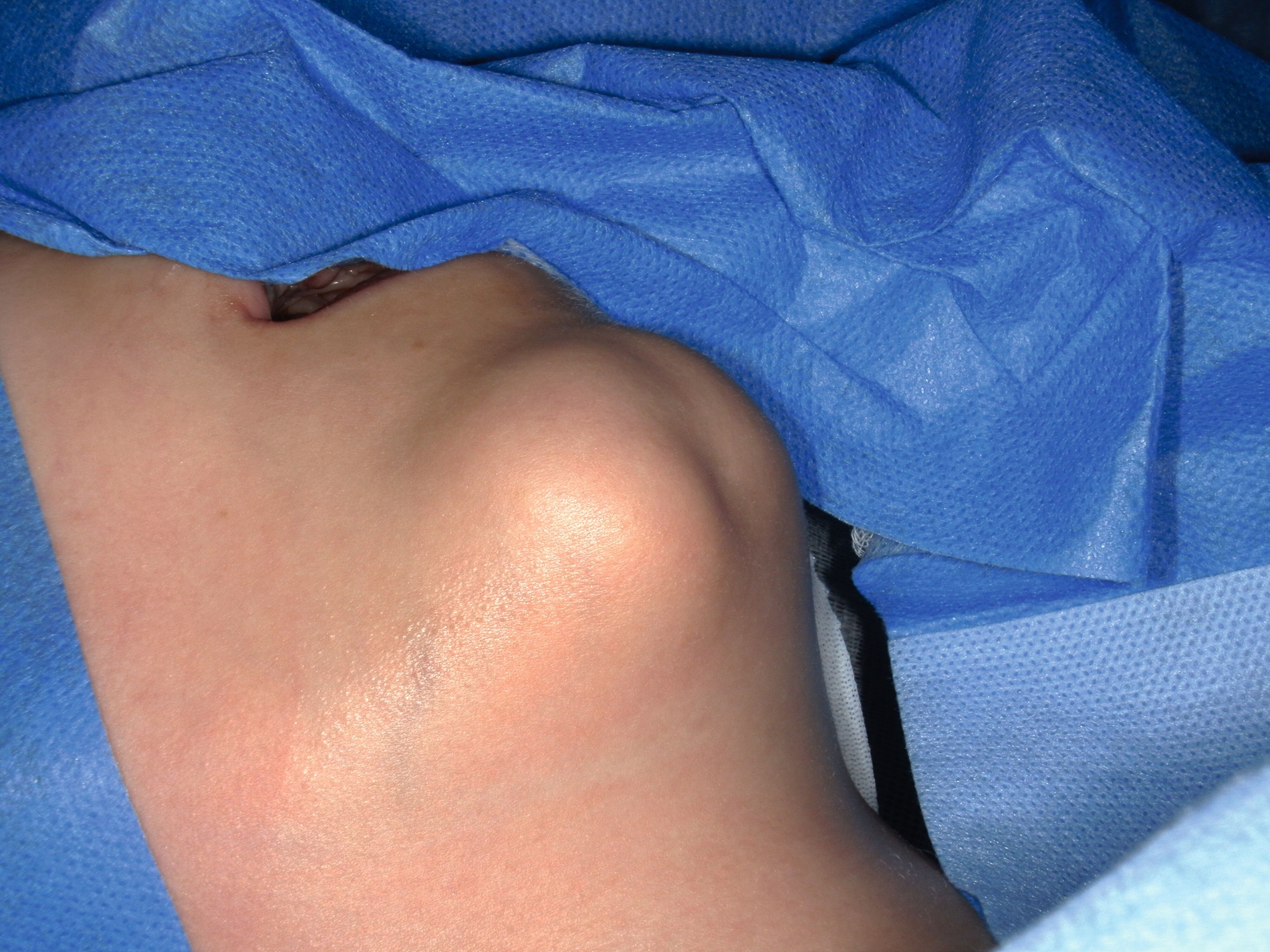
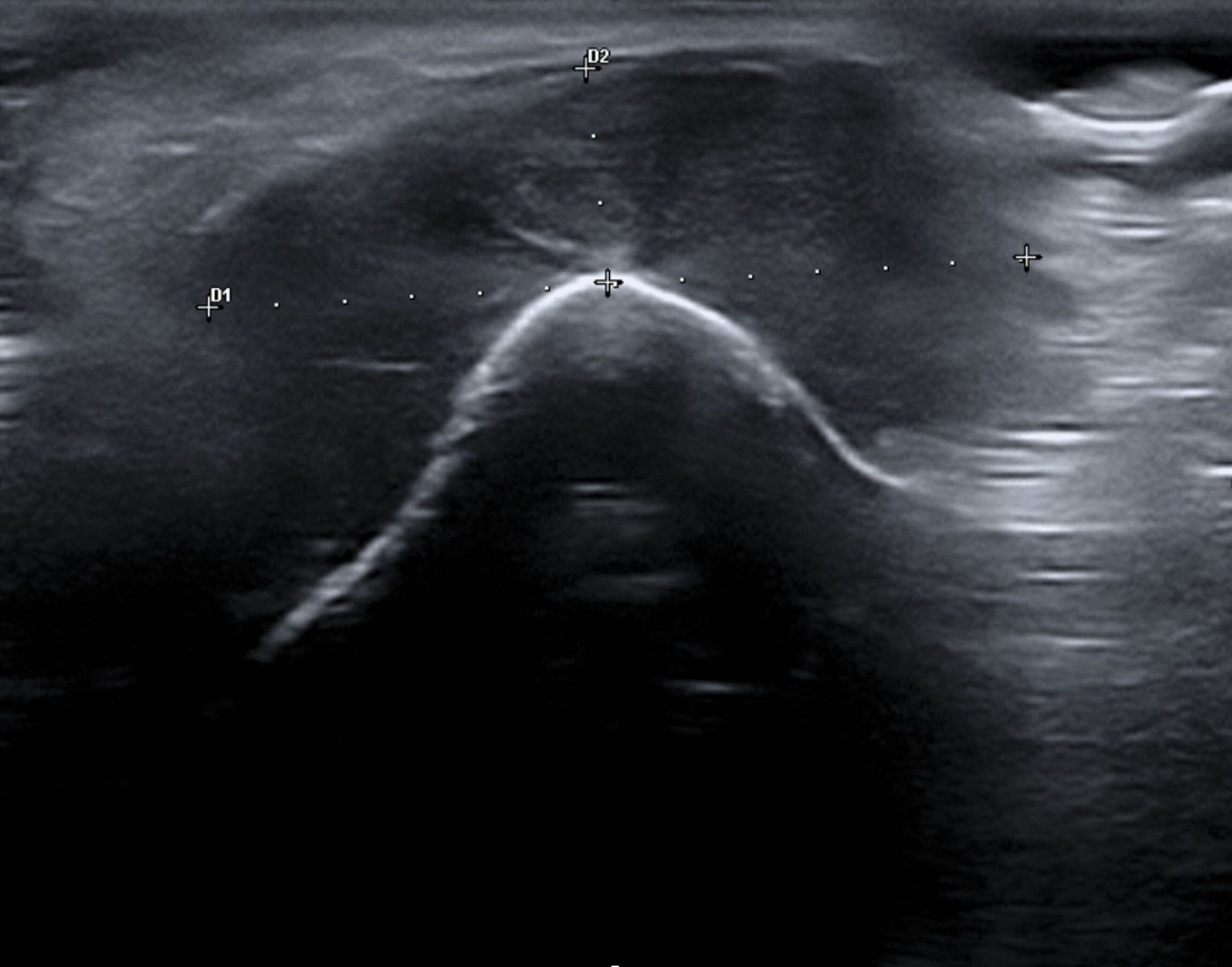
Aufgrund einer Größenprogredienz des Befunds mit inzwischen auch ästhetischer Einschränkung wurde schließlich doch eine Vorstellung bei einem MKG-Chirurgen empfohlen, um die Abklärung der Raumforderung zu initiieren. Klinisch zeigte sich ein derb palpabler, nur teilweise verschieblicher Befund im Bereich des rechten Kinns (Abbildung 1). Weitere Auffälligkeiten oder Hautbefunde kamen nicht zur Darstellung. Sonografisch imponierte ein konzentrisches Muster aus echoarmen und echoreichen Schichten (Abbildung 2).
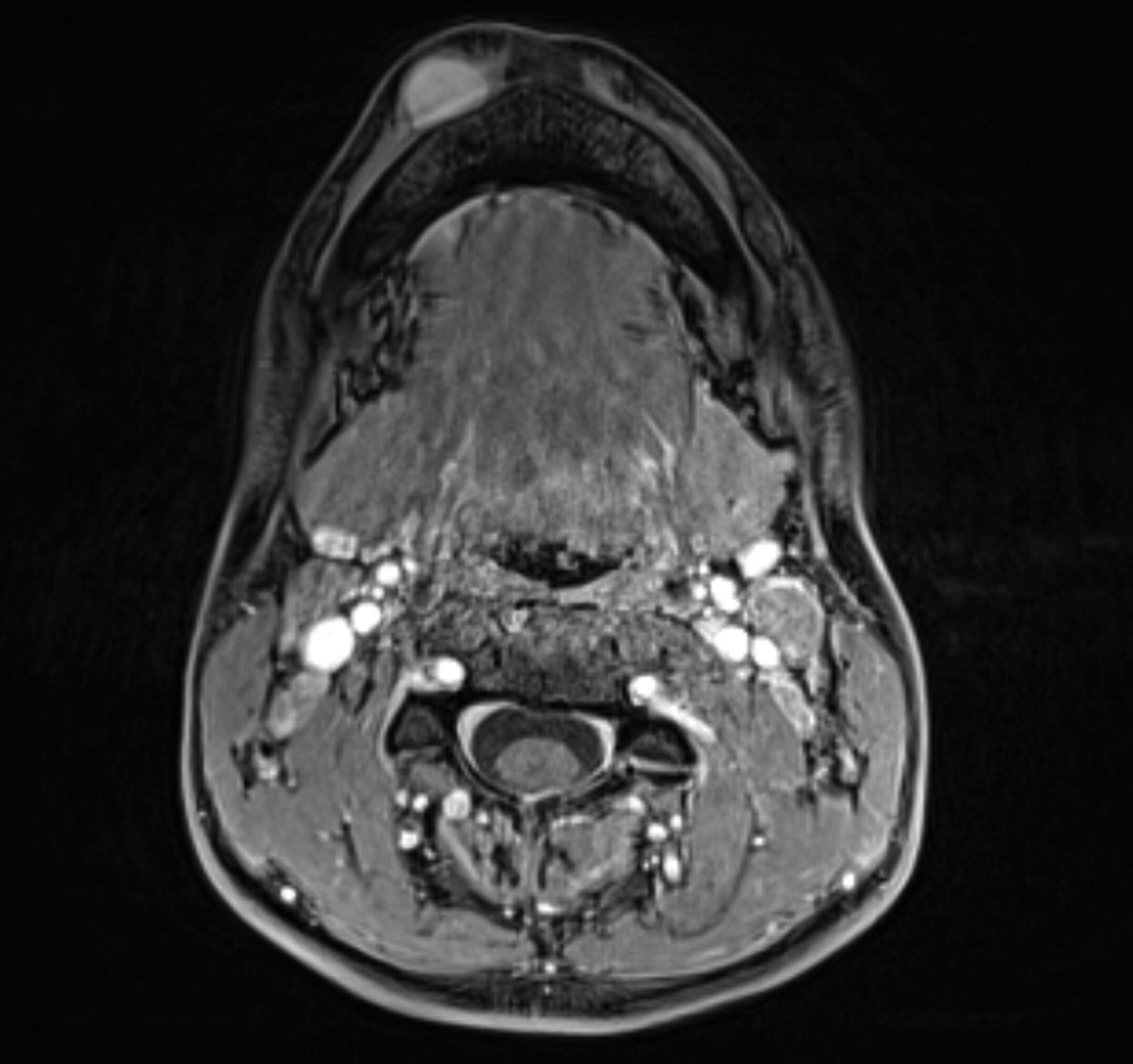
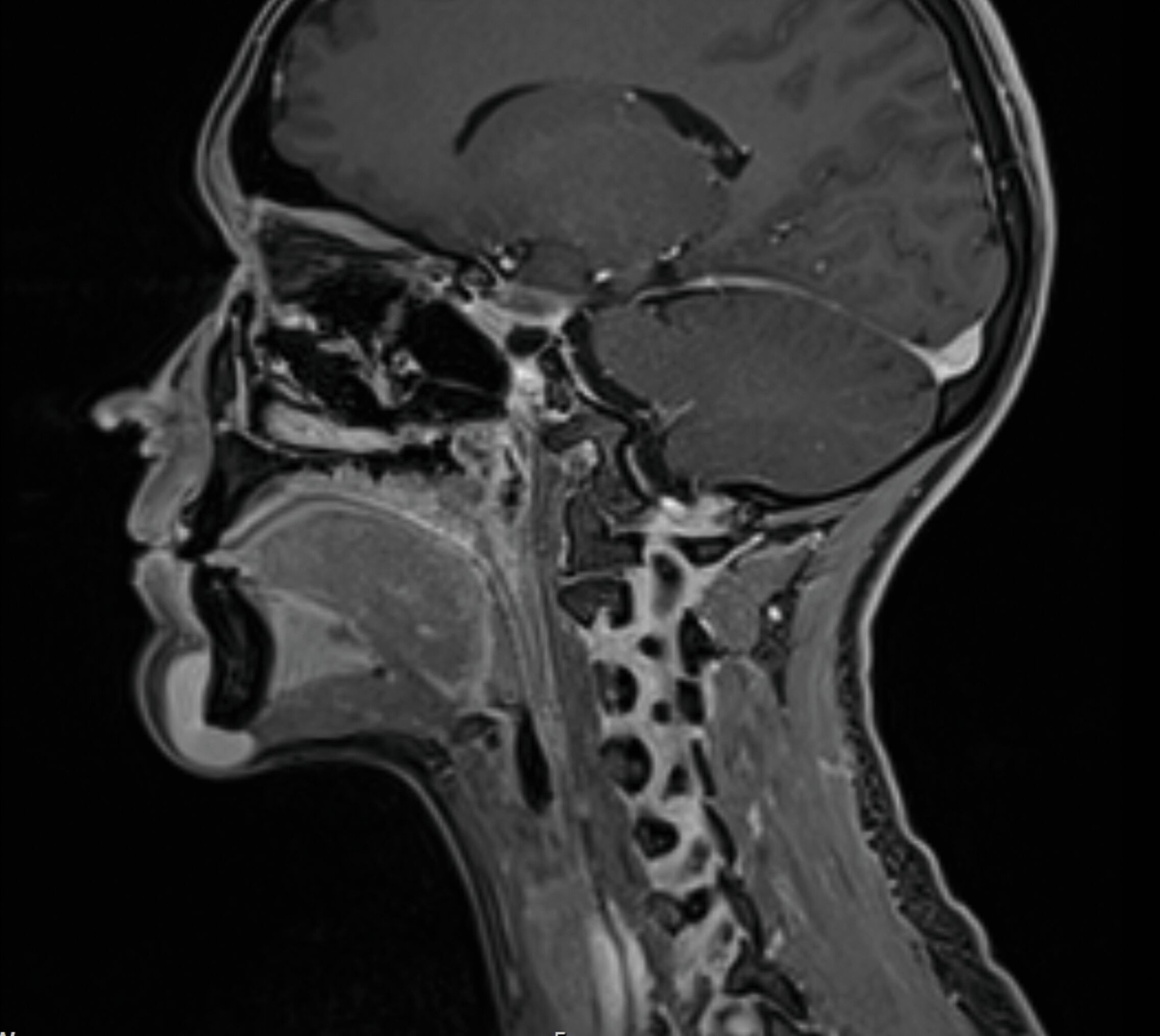
Eine Unterbrechung der Kortikalis oder Abszessformationen ergaben sich nicht. Zur erweiterten Diagnostik erfolgte die Anfertigung einer Magnetresonanztomografie des Kopf-Hals-Bereichs. Hier zeigte sich bildmorphologisch ein zur Muskulatur isointenser Tumor mit deutlicher Kontrastmittelaufnahme, den Unterkiefer basal teilweise umschlingend, jedoch nicht destruierend (Abbildungen 3 und 4).
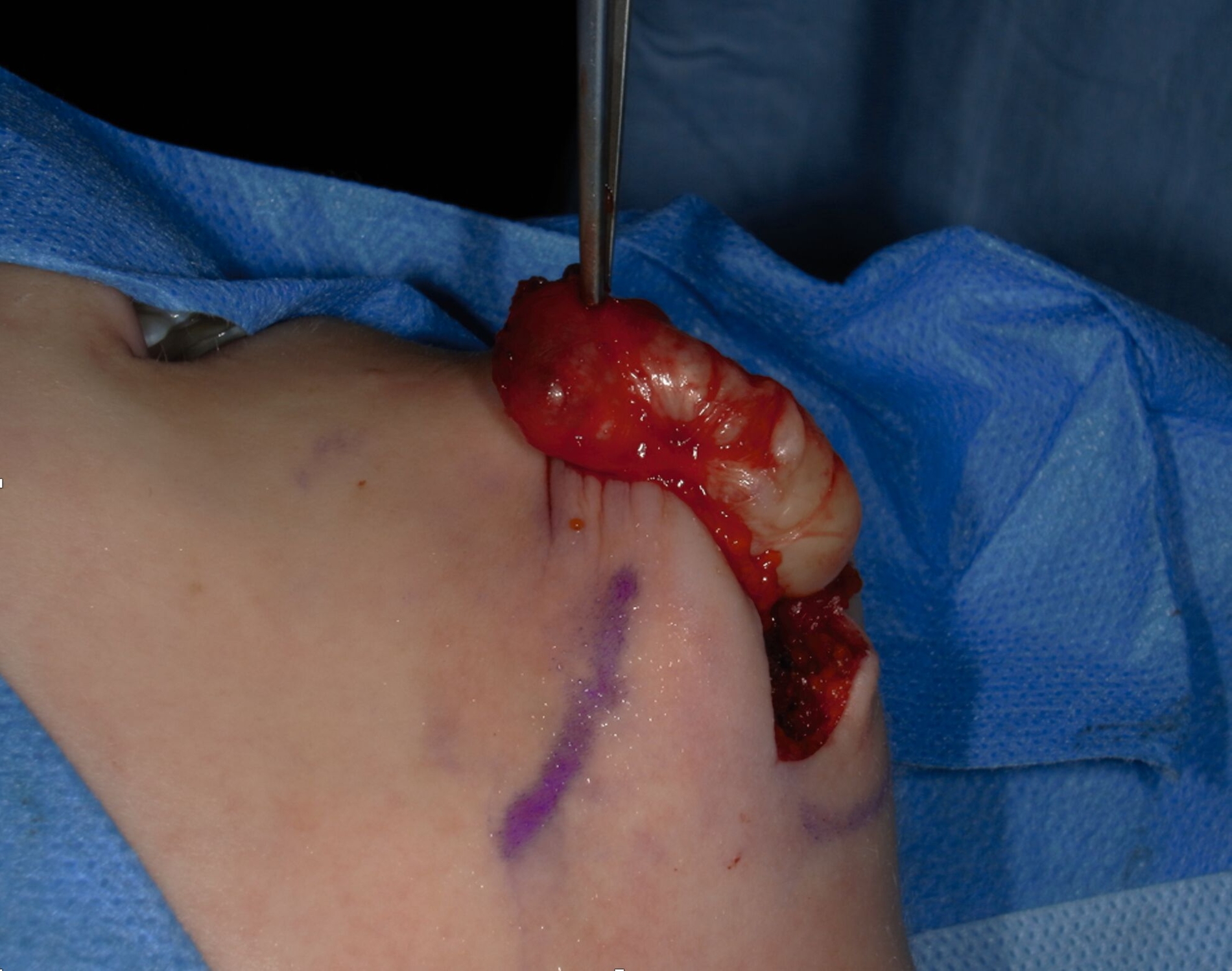
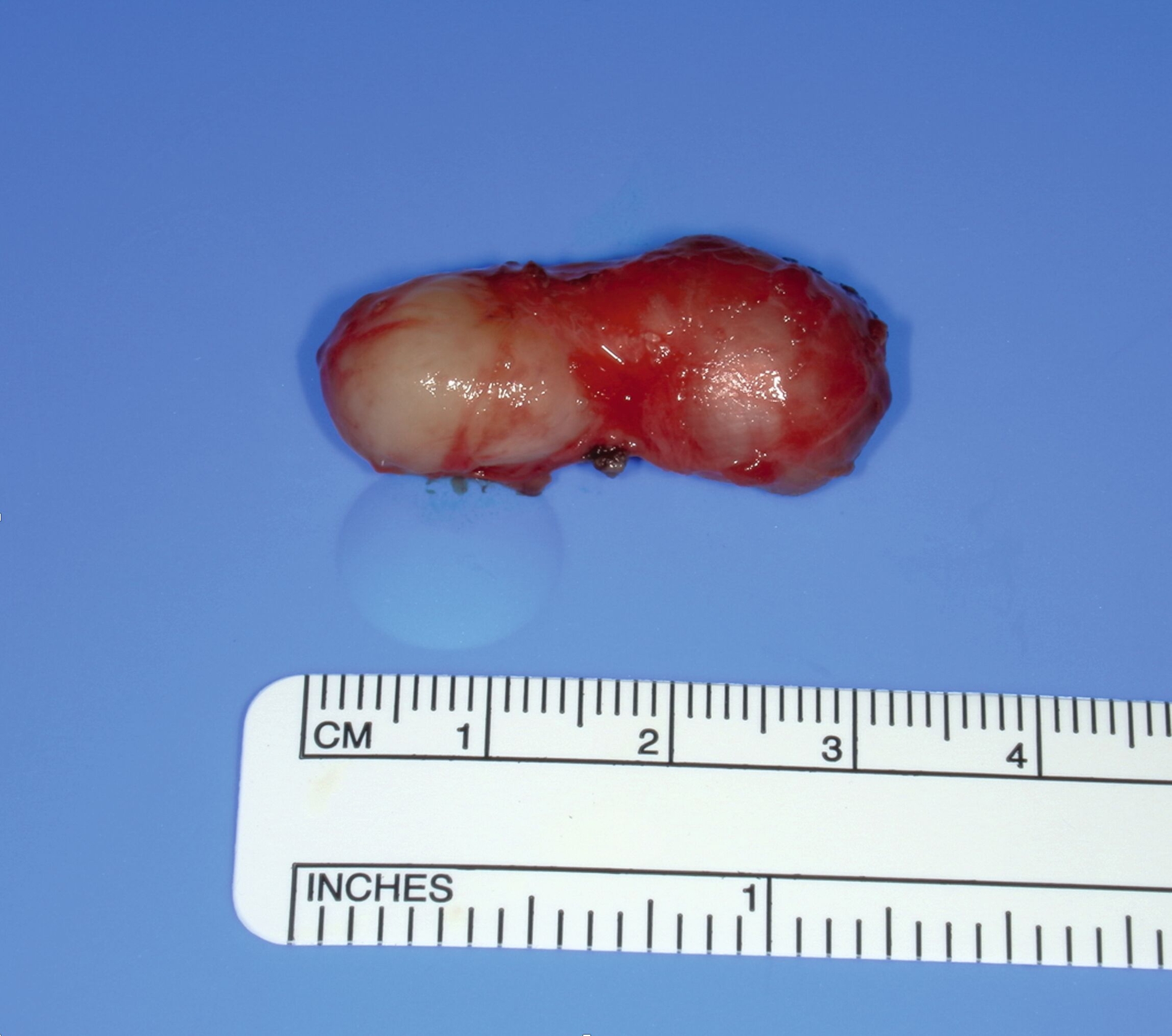
Nach ausführlicher Beratung der Patientin erfolgte in einer gemeinsamen Entscheidung die Terminierung einer Operation zur Entfernung des Befunds. Der Eingriff wurde in Intubationsnarkose durchgeführt. Über einen extraoralen submentalen Zugang konnte der Befund unter Schonung der Nachbarstrukturen dargestellt und in toto entfernt werden (Abbildung 5). Es zeigte sich ein circa 3,5 cm großer, leicht derb palpabler Tumor, der zur weiteren Aufbereitung ans pathologische Institut der Universitätsmedizin entsandt wurde (Abbildung 6). Die Aufbereitung zeigte ein lokalisiertes Neurofibrom.
Der weitere Verlauf gestaltete sich komplikationslos, so dass die Patientin nach einer kurzen stationären Überwachung in die ambulante Weiterbehandlung entlassen werden konnte. Nach Rücksprache mit der humangenetischen Abteilung der Universitätsmedizin Mainz wurde bei einem singulären Neurofibrom auf eine weitere genetische Diagnostik verzichtet.
Diskussion
Neurofibrome sind gutartige Tumore, die von den Nervenscheiden der peripheren Nerven ausgehen. Sie treten häufig singulär oder in Vergesellschaftung mit Neurofibromatose Typ I (von Recklinghausen Neurofibromatose, NF1) auf. Ein vergleichsweise seltenes Auftreten wird in Patienten mit Neurofibromatose Typ II (NF2) beobachtet. Diese zwei Formen der Neurofibromatose unterscheiden sich in ihrem klinischen Erscheinungsbild.
NF1 ist eine autosomal-dominant vererbte Erkrankung mit einigen Hauptkriterien, die unter anderem Café-au-lait-Flecken, Lisch-Knötchen der Iris, axilläres und inguinales Freckling (sommersprossenartige Hyperpigmentierungen) sowie sichtbare kutane und subkutane Neurofibrome umfassen [Tamura, 2021]. Ausgehend von einem Gendefekt auf Chromosom 17 findet sich je nach Penetranz bei den betroffenen Patienten ebenfalls eine Manifestation an inneren Organen oder tiefer gelegenen Nervenplexi. In etwa zehn Prozent der Fälle kann es zur Ausbildung von malignen peripheren Nerventumoren kommen. Die Lebenserwartung von Patienten mit NF1 ist im Vergleich zur Durchschnittsbevölkerung um 15 Jahre herabgesetzt.
NF2 beschreibt ein autosomal-dominant vererbtes Krankheitsbild mit einem Gendefekt auf Chromosom 22 und hoher Penetranz. Patienten mit NF2 entwickeln nahezu alle ein bilaterales Akustikusneurinom. Darüber hinaus finden sich häufig Schwannome an zentralen und peripheren Nerven sowie ein gehäuftes Auftreten von Meningiomen. In der Folge leiden betroffene Patienten häufig unter Hörverlust, Gleichgewichtsstörungen und neuropathischen Störungen. Die Lebenserwartung der Patienten ist reduziert und beträgt im Median 36 Jahre [Godel et al., 2019].
Bei singulär auftretenden Neurofibromen liegt der Altersgipfel zwischen der zweiten und der dritten Lebensdekade, wobei die Geschlechterverteilung gleich ist. Bei einer Manifestation der Erkrankung im Kopf-Hals-Bereich kommt es häufig zu ästhetischen Einschränkungen, weshalb Patienten einen Chirurgen zur Therapie aufsuchen. Man unterscheidet verschiedene Formen des Neurofibroms. Neben dem lokalisierten, meist singulär auftretenden Neurofibrom sind das diffuse und das plexiforme Neurofibrom weitere Formen dieser gutartigen Nerventumoren.
Während das diffuse Neurofibrom vorwiegend bei Kindern und jungen Erwachsenen vorkommt, ist das plexiforme Neurofibrom pathognomonisch für NF1 [Hernandez-Martin et al., 2016]. Plexiforme Neurofibrome können eine maligne Transformation erfahren und sich somit in maligne periphere Nervenscheidentumoren umwandeln.
Histologisch bestehen Neurofibrome vorwiegend aus Schwannzellen, Perineuralzellen, Fibroblasten und Endoneurium [Skovronsky und Oberholtzer, 2004]. Die Läsionen bestehen aus längsovalen und spindelförmigen Zellen. Tumorzellen zeigen in der immunhistologischen Aufbereitung eine schwache Positivität für S-100, Vimentin und fokal auch für CD34. Die immunhistologische Aufbereitung ist ein entscheidender Faktor zum Ausschluss anderer differenzialdiagnostischer, teilweise maligner Tumore wie dem Schwannom oder dem Dermatofibrosarkom. Die chirurgische Therapie mittels Exzision der Tumore stellt derzeit den Goldstandard dar. Leider neigen gerade große Tumore im Bereich eines Nervenplexus zu hohen Rezidivraten, so dass nicht selten ein erneuter Eingriff nötig wird.
Fazit für die Praxis
Neurofibrome sind gutartige Tumore des peripheren Nervensystems.
Die chirurgische Entfernung stellt bei größeren Befunden die Therapie der Wahl dar.
Eine weitere, humangenetische Abklärung zum Ausschluss einer Neurofibromatose kann in bestimmten Fällen indiziert sein.
Derzeit ist eine medikamentöse Therapie der NF1 Gegenstand der Forschung. Eine „Targeted Therapy“ mit gezielter Blockade verschiedener Signalwege könnte die Entstehung von peripheren Nerventumoren unterbinden. Derzeit existiert keine effektive Therapie von NF2. Da entfernte Neurofibrome in diesen Fällen zu häufigen Rezidiven neigen, erfolgt eine chirurgische Therapie nur bei bedrohlichem Wachstum mit möglicher Schädigung anderer anatomischer Strukturen wie beispielsweise bei einer Hirnstammkompression oder bei Gesichtsnervenausfällen.
Literaturliste
Tamura R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int J Mol Sci. 2021;22(11).
Godel T, Baumer P, Farschtschi S, Gugel I, Kronlage M, Hofstadler B, et al. Peripheral nervous system alterations in infant and adult neurofibromatosis type 2. Neurology. 2019;93(6):e590-e8.
Hernandez-Martin A, Duat-Rodriguez A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots and Freckling. Part II. Other Skin Manifestations Characteristic of NF1. NF1 and Cancer. Actas Dermosifiliogr. 2016;107(6):465-73.
Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin N Am. 2004;15(2):157-66.
Dr. med. Philipp Matheis
Kiefer- und Gesichtschirurgie der
Universitätsmedizin Mainz
Augustusplatz 2, 55116 Mainz
philipp.matheis@unimedizin-mainz.de

Univ.-Prof. Dr. Dr. Peer W. Kämmerer
Stellvertr. Klinikdirektor
Klinik und Poliklinik für MKG-Chirurgie und Plastische Operationen,
Universitätsmedizin der Johannes Gutenberg-Universität Mainz
Augustusplatz 3, 55131 Mainz