Parodontitis als Symptom von Syndromerkrankungen

Die Diagnose „aggressive Parodontitis“ für die mit diesen Erkrankungen einhergehenden Parodontitiden ist nicht zutreffend, da die Diagnose aggressive Parodontitis voraussetzt, dass der Patient abgesehen von der Parodontitis gesund ist [Armitage 1999]. In erster Linie gehören zu diesen Syndromerkrankungen angeborene Defekte des zellulären und/oder humoralen Immunsystems sowie andere Erkrankungen, die mit einer gestörten Leukozytenfunktion verbunden sind. Seltener sind frühzeitig beginnende Parodontitiden die Folge von Defekten des parodontalen Bindegewebes.
Trisomie 21
Die Trisomie 21 (Down-Syndrom, Morbus Langdon-Down, numerische autosomale Chromosomenaberration) ist in den meisten Fällen eine klassische Trisomie, das heißt, es ist infolge einer Non-disjunction ein dreifaches Chromosom 21 vorhanden. In wenigen familiär auftretenden Fällen ist das zusätzliche Chromosom 21 oder ein wesentliches Stück davon an ein anderes Autosom angeheftet (Translokation). Beides führt zu einer intra- und extrauterinen Fehlentwicklung fast sämtlicher Gewebe und Organe. Die Inzidenz ist mit dem Alter der Mutter korreliert. Bezogen auf alle Altersklassen beträgt sie 1:700 Lebendgeborene (35- bis 40-jährige Mütter 0,5 bis 1,3 Prozent, 40- bis 45-jährige Mütter 1,3 bis 4,4 Prozent). Das klinische Bild zeigt meist eine erhebliche, aber individuell verschieden entwicklungsfähige geistige Behinderung und eine unterschiedlich ausgeprägte, typische Dysmorphie. Im Kopfbereich imponieren eine Brachyzephalie, Mikrozephalie, lateral-kranial ansteigende Lidachsen, Epikanthus, Hypertelorismus, eine breite Nasenwurzel, tiefsitzende Ohren, eine Unterentwicklung der Kiefer und Zähne (Hypodontie), ein meist offener Mund, vermehrte Speichelsekretion und eine große gefurchte Zunge. Der Schluss der Schädelnähte und der Fontanellen ist verspätet. Weiterhin typisch sind ein rundlicher Minderwuchs, Muskelhypotonie, Cutis laxa, ein tiefstehender Nabel (oft mit Hernie), Vierfingerfurche in den Handflächen, Einwärtskrümmung der Endglieder des fünften Fingers, Fußdeformitäten und ein Herzfehler in 40 bis 60 Prozent der Fälle. In zunehmendem Alter treten überdurchschnittlich häufig Leukämien auf. Es zeigen sich sowohl aggressiv als auch chronisch verlaufende Parodontitiden [Klaus et al. 1987], die ihre Ursache wahrscheinlich in einer Kombination aus genetischen und lokalen Faktoren haben (Abbildungen 1a, b). Zur Entstehung einer aggressiv verlaufenden Parodontitis sollen eine Steigerung der Matrix-Metalloproteinasen( MMP)-2-Aktivität [Komatsu et al. 2001] und eine virale Superinfektion bereits bakteriell infizierter und pathologisch vertiefter Taschen beitragen [Hanookai et al. 2000]. Die Diagnose wird durch den typischen Phänotyp bereits bei der Geburt oder durch pränatale Amniozentese oder Chorionzottenbiopsie gestellt beziehungsweise gesichert. Bezüglich der Prognose: Früher starben 75 Prozent der Patienten vor der Pubertät vor allem infolge einer erhöhten Infektanfälligkeit. Heute werden die Patienten meist älter, 80 Prozent erreichen das 30. Lebensjahr. Bei gezielter, frühzeitig begonnener und individuell angepasster Förderung sind Kinder mit Down-Syndrom lernfähig und sozial gut integrierbar; sie können eine gewisse Selbständigkeit erwerben, die unter Umständen auch den Besuch von Regelschulen erlaubt.
Leukocyte Adhesion Deficiency Syndrome (LADS)
Das Leukocyte Adhesion Deficiency Syndrome (LADS) ist ein seltener angeborener, autosomal-rezessiv vererblicher Defekt der Adhäsionskaskade. Während bei LAD Typ I die Integrinfamilie defekt ist, ist bei LAD Typ II das Selektinsystem betroffen [Etzioni und Tonetti 2000]. Die Patienten leiden unter verschiedenen bakteriellen Infektionen, wie wiederkehrende Hautinfektionen, Otitis media, Septikämie, vermehrte Pusbildung und verzögerte Wundheilung. Diese Symptome werden durch eine persistierende Leukozytose im peripheren Blut begleitet. Das Syndrom geht mit einer sehr aggressiv verlaufenden Parodontitis einher. Durch mangelnde Haftung der Leukozyten an das Endothel und fehlende Einwanderung in das entzündete Gewebe [Meyle 2000] ist die Infektionsabwehr allgemein herabgesetzt. Neben dem Leukozytendefekt leiden die Patienten mit LAD II an Wachstumsstörungen und mentaler Retardierung und zeigen den seltenen Bombay-Blutgruppentyp. Prognose: Bei intensiver Antibiotikatherapie wird das Erwachsenenalter erreicht. Eine weitere Therapiemöglichkeit ist eine Knochenmarkstransplantation.
Papillon-Lefèvre-Syndrom
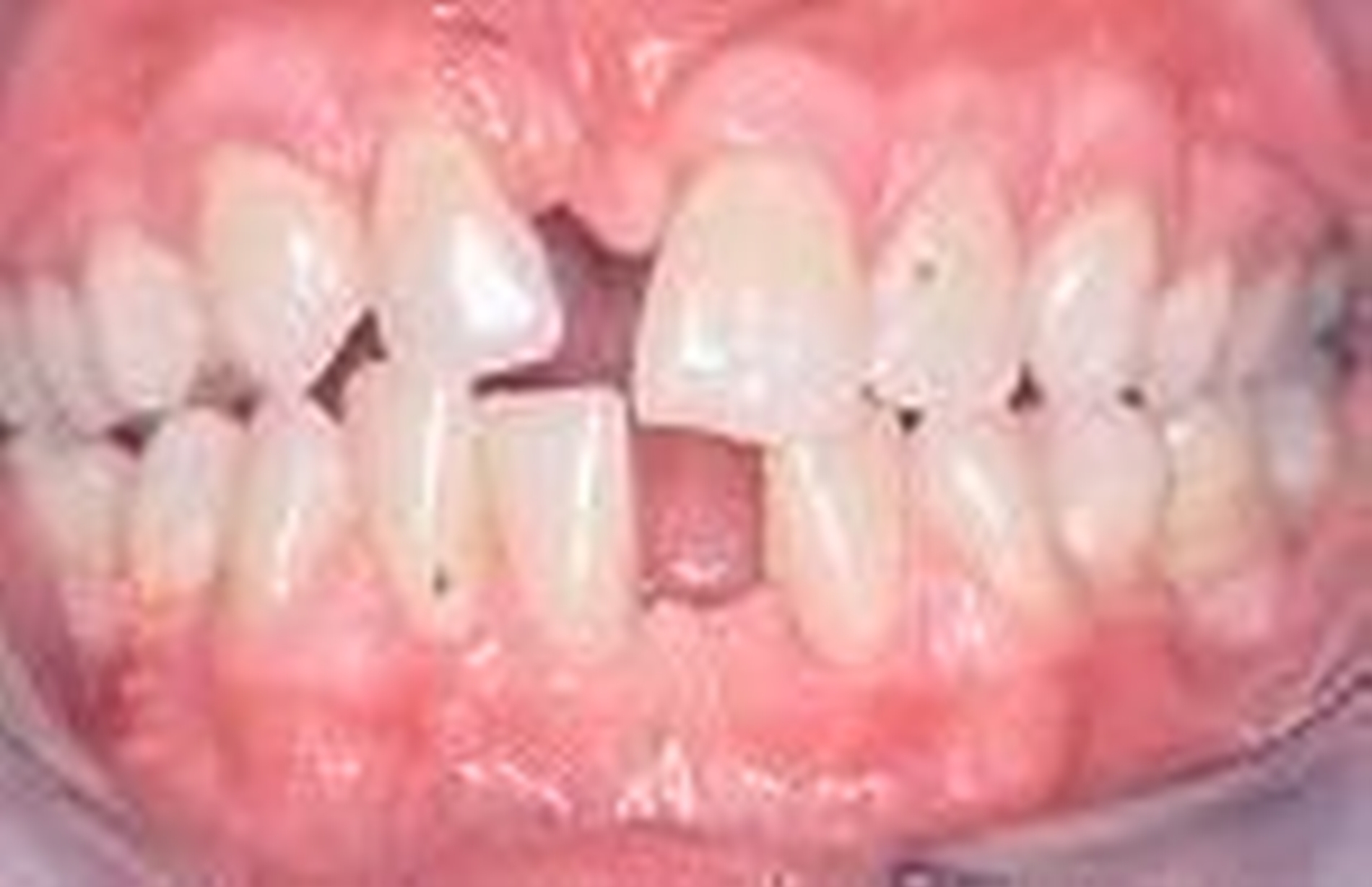
Das Papillon-Lefèvre-Syndrom beschreibt eine seltene, autosomal-rezessiv erbliche Palmoplantarkeratose (Abbildung 2a, b) mit einer früh einsetzenden generalisierten Parodontitis (Abbildung 2c, d), die sowohl die erste als auch die zweite Dentition betrifft [Fischer et al. 1997, Papillon und Lefèvre 1924]. Die Häufigkeit beträgt eins bis vier : 1 000 000 [Preus und Gjermo 1987]. Neuerdings wurde bei betroffenen Patienten eine Mutation des Kathepsin-C-Gens nachgewiesen [Hart et al. 2000, Toomes et al. 1999]. Untersuchungen der neutrophilen Granulozyten haben in manchen Fällen gestörte Zellfunktionen, wie eine verminderte Motilität, Chemotaxis und Phagozytose sowie eine verminderte Produktion von Sauerstoffradikalen gezeigt, die sich in manchen Fällen nach der therapeutischen Intervention normalisierten [Bimstein et al. 1990, Bullon et al. 1993, Firatli et al. 1996, Ghaffar et al. 1999, Liu et al. 2000]. In der subgingivalen Plaque konnten vermehrt anaerobe gramnegative Keime undActinobacillus actinomycetemcomitansnachgewiesen werden [Clerehugh et al. 1996, Lundgren et al. 1988, Velazco et al. 1999]. Die Symptomatik umfasst psoriasiforme Hyperkeratosen an Ellenbogen und Knien, Nageldystrophie und gehäufte bakterielle Infekte. Eine Therapie der Hautveränderungen erfolgt mit Retinoiden [Bergman und Friedman-Birnbaum 1988, Lundgren et al. 1996]. Für die parodontale Therapie erscheint die Elimination vonActinobacillus actinomycetemcomitansfür den Therapieerfolg von entscheidender Bedeutung zu sein [Eickholz et al. 2001, Kressin et al. 1995, Rüdiger et al. 1999].
Haim-Munk-Syndrom
Das Haim-Munk-Syndrom ist ein weiteres autosomal-rezessiv vererbbares Syndrom, das nur in einer jüdischen Bevölkerungsgruppe in Indien beschrieben wurde. Mit palmoplantaren Hyperkeratosen und aggressiv verlaufender Parodontitis ist es dem Papillon-Lefèvre-Syndrom ähnlich. Es zeigen sich alle Mutationen des Kathepsin-CGens [Hart et al. 2000]. Weitere Symptome sind Onychogrypose, Arachnodaktylie und Akroosteolyse.
Chediak-Higashi-Syndrom
Das Chediak-Higashi-Syndrom ist eine seltene autosomal-rezessiv erbliche Stoffwechselanomalie mit Störungen der Hautpigmentierung und der zellulären Immunität. Das Krankheitsbild wird vermehrt in jüdischen Bevölkerungsgruppen beobachtet. Als Symptome zeigen sich mit Manifestation im Kindesalter eine Disposition zu rezidivierenden Infektionen, eine allgemeine Hypopigmentation, ein partieller Albinismus und ein Albinismus fundi occuli, eine Hepatosplenomegalie und eine Lymphadenopathie. Typischerweise geht es mit einer aggressiv verlaufenden Parodontitis einher. Der erhöhten Infektanfälligkeit liegen abnorme lysosomale Riesengranula mit einer gestörten Degranulation zu Grunde [Delcourt-Debruyne et al. 2000]. Bei der Diagnostik zeigen sich im peripheren Blutbild eine Granulationsanomalie der Leukozyten und Lymphozyten (Riesengranula) und plasmatische Einschlusskörperchen in den myeloischen Zellen im Knochenmark. Pränatal sind eine fetale Blutuntersuchung und eine Haut-Haar-Biopsie möglich. Die Therapie besteht in einer Knochenmarkstransplantation. Die Prognose ist auf Grund der Disposition zu septischen Prozessen im Kindesalter ungünstig. Die meisten Kinder sterben vor Erreichen des zehnten Lebensjahres an Infekten oder malignen Tumoren.
Histiozytose-Syndrom
Die Langerhans-Zellhistiozytose (LCH) ist ein Oberbegriff für reaktiv-proliferative Erkrankungen mit Vermehrung von Histiozyten des Langerhans-Zellphänotyps in den suprabasalen Schichten der Haut und der Schleimhäute [Chu et al. 1987, Favara et al. 1997]. Die Ätiologie ist unklar, möglicherweise entsteht die LCH durch eine atypische Immunantwort oder es handelt sich um eine Autoimmunkrankheit. Aktuelle Untersuchungen belegen, dass es sich auch um eine klonale proliferative Erkrankung handeln könnte. Eine Manifestation der LCH tritt meist vor dem 30. Lebensjahr auf, wobei intraorale Läsionen häufig anzutreffen sind (Abbildungen 3a, b) [Hartman 1980]. Während extraoral im Bereich der Axillen und prästernal häufig krustöse Hautveränderungen imponieren, treten intraoral oft ulzerierende, nekrotisierende parodontale Läsionen auf [Hartman 1980, Sedano et al. 1969, Sigala et al. 1972]. Röntgenologisch findet sich häufig das Bild der „schwimmenden Zähne“ mit multiplen ausgeprägten radikulären Osteolysen [Yu et al. 1995, Pringle et al. 1992, Sedano et al. 1969]. Es gibt drei sich zum Teil überlappende Formen, welche früher als eosinophiles Granulom, Letterer-Siwe-Krankheit und Hand-Schüller-Christian-Krankheit bezeichnet und unter dem Sammelbegriff Histiozytose X auf Grund der unklaren Ätiologie zusammengefasst wurden [Lichtenstein 1953]. Die Diagnose wird histologisch, immunhistochemisch durch den Nachweis von CD1a-Antigen und S-100 Protein auf den Membranen der Langerhans-Zellen und elektronenmikroskopisch über Birbeck-Granula in Langerhans-Zellen gesichert [DiFranko et al. 1985, Ide et al. 1984].
Glykogenspeicher-Syndrome
Die Glykogenspeicher-Syndrome (Glykogenosen) sind autosomal-rezessiv vererbte Erkrankungen des Glykogenabbaus beziehungsweise der Glykogensynthese mit pathologisch gesteigerter Glykogenspeicherung in vielen Organen (Leber, Nieren, Herz, Muskulatur, ZNS). Es gibt je nach vorhandenem Enzymdefekt verschiedene Formen beziehungsweise Typen. Die Symptome reichen von einer Hepatomegalie, Minderwuchs, Hypoglykämie, Neutropenie, Kardiomyopathie, Muskelhypotonie bis zu einer progressiven Hirndegeneration. Glykogenose Typ I ist häufig mit Neutropenie und verminderter neutrophiler Chemotaxis assoziiert [Hara et al. 1987, Koven et al. 1986]. Eine pränatale Diagnostik ist nicht bei allen Formen möglich. Bei den Formen mit Hypoglykämie ist eine Therapie mit einer Diät auf der Basis ungekochter Stärke erfolgreich.
Infantile genetische Agranulozytose
Die Infantile genetische Agranulozytose (Kostmann-Syndrom, chronic granulomatous disease) ist die häufigste angeborene, sich früh manifestierende hochgradige Granulozytenfunktionsstörung mit Auftreten akuter, lebensbedrohlicher bakterieller undmykotischer Infektionen (schwere und lang anhaltende Pyodermien, Dermatitiden im Nasen-Mund-Bereich, ekzematöse Läsionen, Lymphknotenvereiterungen und septische Bakterienabsiedelungen im Knochen, Darm, Leber und Lunge) [Carlsson und Fasth 2001]. Die häufigsten Erreger sind katalasepositive Bakterien, wie Staphylococcus aureus, Klebsiellen und Aspergillus-Spezies. Orale Manifestationen sind chronische Ulzerationen des harten Gaumens und der Alveolarmukosa, Gingivitiden und in der Pubertät beginnende aggressiv verlaufende Parodontitiden. Die Ursachen liegen in einem gestörten Phagozytosevorgang der CGD-Granulozyten. Die Erkrankung betrifft Jungen vier bis fünf mal häufiger als Mädchen. Es liegt ein X-chromosomal gebundener Erbgang vor. Die Therapie besteht aus einer lebenslangen Substitution mit gentechnisch hergestelltem Granulozyten-Wachstumsfaktor (G-CSF) oder bei einem passenden Spender auch aus einer Knochenmarkstransplantation.
Ehlers-Danlos-Syndrom
Ehlers-Danlos-Syndrom (Fibrodysplasia elastica generalisata congenita) ist eine Bezeichnung für eine Gruppe erblicher Krankheitsbilder mit Kollagendysplasie, die sich nach biochemischen, genetischen und klinischen Kriterien in zehn verschiedene Typen aufgliedert. Die Häufigkeit (in England) beträgt 1:150 000. Die Ätiologie ist je nach Typ ein autosomal-dominanter, -rezessiver oder Xchromosomaler Erbmodus. Es gibt entsprechend unterschiedliche pathochemische Mechanismen der gestörten Kollagenfibrillogenese. Die Symptome bestehen je nach Typ aus einer unterschiedlichen Symptomenkonstellation und -schwere mit Hyperelastizität und erhöhter Vulnerabilität sowie Wundheilungsstörungen der Haut, Überstreckbarkeit der Gelenke mit Luxationsneigung, Augenanomalien (zum Beispiel Myopie, Linsenektopie, blauen Skleren, Neigung zu Netzhautblutungen), Disposition zu vasogener Hämorrhagie, Aneurysma dissecans und Arterienrupturen, verstärkten Nachblutungen bei operativen Eingriffen und einer erhöhten Frühgeborenenrate. Vor allem die Typen IV, VII und IX mit Kollagenase-, Prokollagenpeptidase- und Kollagendefekten manifestieren eine aggressiv verlaufende Parodontitis [Cunniff und Williamson-Kruse 1995, Karrer et al. 2000]. Die Diagnostik erfolgt mittels biochemischem Nachweis spezifischer Enzymdefekte. Pränatal ist bei einigen Typen eine Diagnosesicherung durch Chorionzottenbiopsie oder Amniozentese möglich.
Hypophosphatasie
Die Hypophosphatasie (Rathbun-Syndrom) ist eine eher seltene, autosomal-rezessiv vererbte, angeborene Stoffwechselstörung mit verminderter Aktivität der alkalischen Phosphatase [Watanabe et al. 1999]. Dies führt zu einer gestörten Mineralisation des Skeletts (Kraniosynostose, Thoraxdeformitäten) und zu einer vermehrten Ausscheidung von Phosphoethanolamin im Harn. Eine vorzeitige Exfoliation der Milchzähne durch eine aggressiv verlaufende Parodontitis wird als konstantes Charakteristikum derjenigen Fälle beschrieben, die infolge milder klinischer Erscheinungen nicht bereits während der ersten zwei Lebensjahre diagnostiziert werden (Abbildungen 4a, b). Es liegen dentale Abnormalitäten, wie abnorme Schmelz-, Dentin- und Wurzelzementbildung, vor [Watanabe et al. 1993]. Formen: Letaler kongenitaler Typ mit Osteopenie des gesamten Skeletts; Tod bereits in utero oder in den ersten Lebenstagen. Kindlicher Typ mit autosomal-rezessivem Erbgang, schwerem Verlauf und ungünstiger Prognose. Erwachsenen-Typ mit autosomal-dominantem Erbgang und leichten Rachitis-ähnlichen Symptomen.
Schlussbemerkung
Die meisten der aufgeführten Syndromerkrankungen sind sehr selten und zum Teil wie die aggressiv verlaufende Parodontitis beim Chediak-Higashi-Syndrom primär zahnärztlich parodontologisch gar nicht therapierbar. In vielen Fällen wie bei der mit der Trisomie 21 einhergehenden Parodontitis kommt es aber darauf an, diese frühzeitig zu erkennen oder, noch besser, die betreffenden Patienten rechtzeitig präventiv in der Praxis des Hauszahnarztes zu betreuen. Die frühzeitige Exfoliation der Milchzähne sollte vom Zahnarzt nicht achselzuckend registriert werden, sondern Anlass dazu geben, nach Symptomen für Syndromerkrankungen zu suchen (wie Hauteffloreszenzen bei Papillon-Lefèvre-Syndrom) oder bei Durchbruch der bleibenden Zähne frühzeitig auch parodontale Parameter zu erheben, zum Beispiel mit dem Parodontalen Screening Index (PSI). So können behandelbare parodontale Manifestationen frühzeitig erkannt und die Patienten rechtzeitig an Spezialisten beziehungsweise Fachzahnärzte für Parodontologie überwiesen werden.
Korrespondenzadresse:Dr. Filip Klein, Sektion ParodontologiePoliklinik für ZahnerhaltungskundeKlinik für Mund-, Zahn- und KieferkrankheitenUniversitätsklinikum HeidelbergIm Neuenheimer Feld 40069120 Heidelberg
\n
Syndrom
Defekt
Verlauf der Parodontitis/Zustand des Parodonts
Erbgang
\n
\n
Trisomie 21
multiple
Chronisch und aggressiv
\n
Leukocyte Adhesion
Deficiency Syndrome
Neutrophile
Granulozyten
Aggressiv
Autosomal-rezessiv
\n
Papillon-Lefèvre- Syndrom
Keratin oder Epithel, Kathepsin C
Aggressiv
Autosomal-rezessiv
\n
Chediak-Higashi Syndrom
Neutrophile
Granulozyten
Aggressiv
Autosomal-rezessiv
\n
Ehlers-Danlos-Syndrom
(Typen IV, VII, IX)
Kollagen Typ III, Prokollagen- peptidase, Kollagen
Aggressiv/
fragile Gewebe
Autosomal- dominant,
\n
-reszessiv bzw. X-chromosomal
\n
Hypophosphatasie
(Rathbun-Syndrom)
Alkalische Phosphatase
Aggressiv/ schlecht mineralisierter
\n
Knochen und Zement
Autosomal-rezessiv
\n
\n

