Angioödeme / Quincke-Ödem
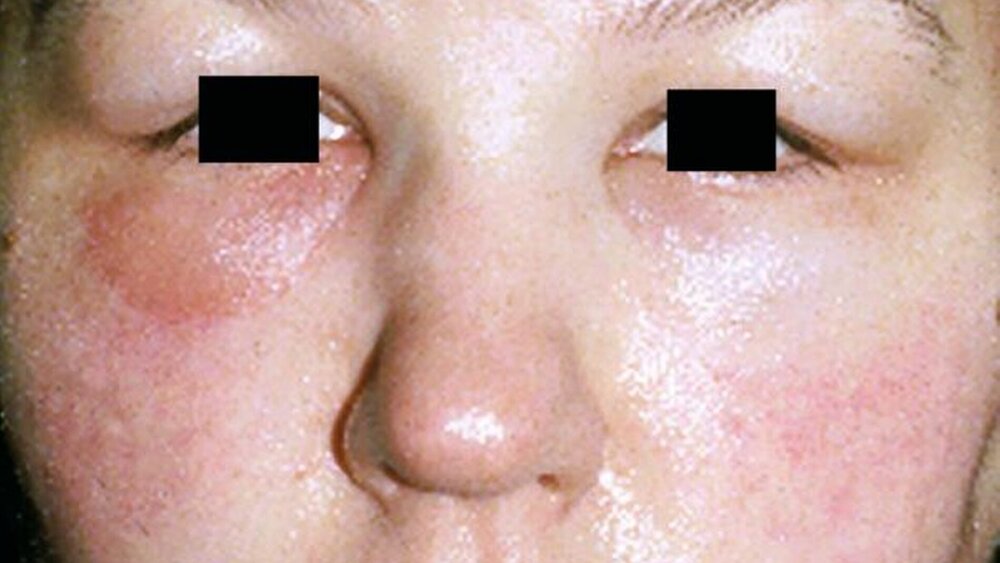

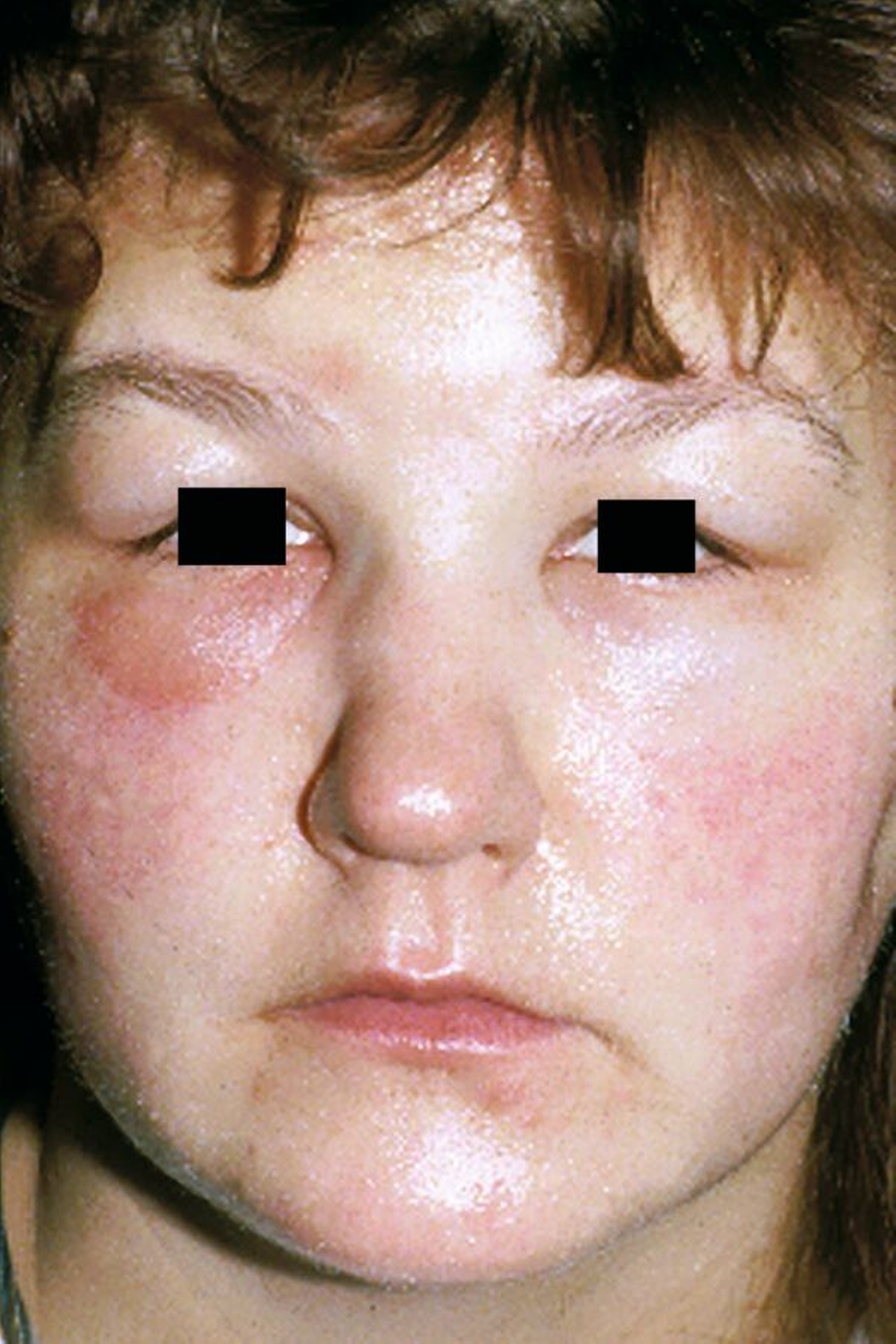
Primär schmerzlose, sich innerhalb von Minuten entwickelnde Schwellungen sind das charakteristische Symptom eines Angioödems – eine Störung, die synonym als Quincke-Ödem bezeichnet wird oder gelegentlich auch noch mit dem veralteten Begriff angioneurotisches Ödem. Die Schwellungen treten in der Regel im Gesichtsbereich auf, betroffen sind zumeist die Augenlider, die Lippen und die Wangen. Nicht selten schwillt aber auch das Gewebe der Zunge oder der Atemwege und insbesondere die Glottis. Es kann dann zur massiven Atemnot bis hin zu einem lebensbedrohlichen Zustand kommen.
Symptome über die Schwellung hinaus
Unabhängig davon klagen die Patienten über Spannungsgefühle in der betroffenen Hautpartie und gelegentlich auch über Juckreiz sowie Schluckbeschwerden, Heiserkeit und eine veränderte Stimme. Die Schwellung stellt oft eine starke Belastung dar und das nicht nur wegen der möglichen Konsequenzen wie etwa der Atemnot. Denn vor allem, wenn das Gesicht betroffen ist, drohen erhebliche Entstellungen. Die Schwellungen, die für Stunden, aber auch über Tage anhalten können, betreffen zudem nicht selten auch den Genitalbereich und/oder den unteren Gastrointestinaltrakt, was unspezifische gastrointestinale Beschwerden wie Leibschmerzen sowie Übelkeit, Erbrechen und einen Aszites nach sich ziehen kann.
Das Angioödem entwickelt sich Histaminvermittelt als allergisch bedingte Reaktion, als Unverträglichkeitsreaktion und damit als Nebenwirkung auf eine Arzneimittel-Einnahme oder im Zusammenhang mit einem angeborenen C1-Esterase-Inhibitor-Mangel, eine Erkrankung, die als hereditäres Angioödem bezeichnet wird.
Bekannt schon seit dem Mittelalter
Angioödeme sind keine „Erfindung“ der modernen Medizin, sondern schon seit dem Mittelalter bekannt: Die Krankheitsberichte gehen bis ins 16. Jahrhundert zurück, wobei aber die genaue klinische Beschreibung des Syndroms erst 1843 erfolgte. Der Begriff des „angioneurotischen Syndroms“ wurde schließlich in den 80er-Jahren des 19. Jahrhunderts durch den deutschen Arzt Heinrich Irenäus Quincke geprägt, was die Bezeichnung der Erkrankung als Quincke-Ödem erklärt. 1888 wurde die erbliche Form des Angioödems beschrieben und schließlich 1917 dessen autosomal dominanter Erbgang.
Ursachen der Schwellung
Die Ursachen der Schwellung der Subkutis respektive der Submukosa sind vielfältig. Die Reaktion kann auf immunologische Prozesse zurückgehen und durch die Ablagerung von Immunkomplexen bedingt sein. Sie steht nicht selten im Zusammenhang mit einer Urtikaria, wobei Literaturberichten zufolge 50 bis 80 Prozent der Patienten mit Nesselsucht Histamin-vermittelte Angioödeme entwickeln. Eine Nesselsucht aber bekommen rund 25 Prozent der Menschen mindestens einmal in ihrem Leben.
Ein Angioödem kann außerdem als Unverträglichkeitsreaktion nach der Einnahme von Medikamenten auftreten, wobei eine solche Nebenwirkung vor allem bei den ACE-Hemmern beschrieben ist und – in jedoch deutlich geringerem Ausmaß – auch bei den modernen Angiotensin-2-Rezeptorblockern. Die Störung kann aber auch idiopathisch und durch physikalische Auslöser wie Wärme, Kälte oder Druck getriggert sein.
Das hereditäre Angioödem (HAE) stellt eine Sonderform des Angioödems dar und beruht direkt auf einem Defekt des C1-Esterase-Inhibitors.
Verursacht wird die Gewebeschwellung beim Angioödem durch den Mediator Bradykinin, einen potenten Vasodilatator, der neben der Gefäßerweiterung auch eine erhöhte vaskuläre Permeabilität auslöst. Die Substanz bewirkt eine rasche Akkumulation von Flüssigkeit im Interstitium, wobei sich die resultierende Schwellung im Gesichtsbereich, also dort, wo nur wenig stützendes Bindegewebe vorhanden ist, besonders rasch entwickelt. Beim HAE wird die Bradykinin-Aktivierung durch den C1-Esterase- Inhibitor-Mangel verursacht, bei den Medikamenten-induzierten Krankheitsfällen ist eine zum Beispiel durch einen ACE-Hemmer blockierte Funktion der Kininase II die Grundlage, da damit der Abbau von Bradykinin gehemmt wird.
Diagnostik schon per Augenschein
Infolge der Schwellung lässt sich das Angioödem in aller Regel schon per Augenschein diagnostisch fassen. Wichtig ist eine genaue Anamnese mit entsprechender Familienanamnese, um die auslösende Situation zu eruieren. So weist beispielsweise eine familiäre Häufung der Probleme direkt auf ein hereditäres Angioödem hin.
Es ist anamnestisch unbedingt auch nach der Einnahme von Medikamenten zu fragen und das auch nach Medikamenten, die bereits seit Längerem eingenommen werden. Denn ein Angioödem kann auch noch als Nebenwirkung eines ACE-Hemmers auftreten, wenn dieser bereits längere Zeit regelmäßig geschluckt wird.
Je nach dem Ergebnis der Anamnese können weiterführende Untersuchungen wie etwa Allergietests notwendig sein oder auch eine Serumbestimmung des C1-Esterase- Inhibitors.
Behandlung des Angioödems
Die wohl wichtigste Maßnahme beim Angioödem ist es, die auslösenden Trigger zu beheben, was allerdings nur bedingt möglich ist. Ist die Störung als Komplikation auf eine Medikamenteneinnahme zu eruieren, so muss das jeweilige Arzneimittel unverzüglich abgesetzt werden, und es ist gegebenenfalls eine andere Medikation zu erwägen. Die Patienten müssen außerdem gut über die Störung aufgeklärt werden, da es bei erneuter Einnahme entsprechender Wirkstoffe, die zum Beispiel bei der Behandlung des Bluthochdrucks weit verbreitet sind, wiederum zu einem Angioödem und potenziell lebensbedrohlichen Komplikationen kommen kann.
Ist das Angioödem Histamin-vermittelt, so wird es üblicherweise durch Antihistaminika und Glukokortikoide behandelt. Die Behandlung richtet sich generell nach der Schwere der Komplikation. So kann bei allergisch vermittelten Reaktionen im Notfall auch eine Adrenalin-Injektion notwendig werden und gegebenenfalls sogar eine endotracheale Intubation und künstliche Beatmung oder im Fall des Falles sogar eine Tracheostomie.
Das hereditäre Angioödem
Das HAE als erblich bedingte Sonderform des Angioödems beruht auf einem Mangel des Proteins C1-Esterase-Inhibitor (C1-INH) infolge einer Mutation auf dem für dieses Protein kodierende Gen auf Chromosom 11. Inzwischen sind mehr als 200 verschiedene Mutationen bekannt, was die zum Teil unterschiedliche Ausprägung des Krankheitsbildes erklärt. Die Erkrankung lässt sich meist über mehrere Generationen in den betroffenen Familien verfolgen. Bei jedem vierten Patienten aber bildet sich die Mutation spontan und es besteht eine negative Familienanamnese.
C1-INH, ein Serinprotease-Inhibitor, gehört zu den Faktoren des Komplementsystems. Es handelt sich konkret um einen zentralen Inhibitor des Kallikrein-Kinin-Systems, der als Regulationsprotein am Beginn der Komplementkaskade steht. Fehlt C1-INH, so kommt es zu einer unkontrollierten Aktivierung des klassischen Weges der Komplementkaskade mit verstärktem Verbrauch der übrigen Faktoren und vermehrter Bildung des Mediators Bradykinin. Beim Fehlen des Inhibitors sind ferner die Fibrinolyse wie auch die intrinsische Gerinnungskaskade in ihrer Regulation beeinträchtigt. Dadurch ist während einer akuten HAE-Attacke stets auch von einer Aktivierung der Blutgerinnung auszugehen, da C1-INH konkret den Faktor XIIa und auch den Faktor XIa sowie das Thrombin hemmt. Trotz der Aktivierung dieser Systeme wurde bei Patienten mit HAE aber kein erhöhtes Thromboserisiko und auch keine verstärkte Blutungsneigung festgestellt.
Beim HAE werden verschiedene Krankheitsformen unterschieden:
• der HAE Typ 1, der bei 85 Prozent der Patienten vorliegt, und bei dem verminderte C1-INH-Spiegel nachzuweisen sind,
• der HAE Typ 2, den 15 Prozent der Patienten aufweisen, und der eine beeinträchtigte Funktion des Faktors beschreibt,
• der HAE Typ 3, bei dem keine eindeutige Anomalie des C1-INH zu finden ist, der aber offenbar X-chromosomal dominant vererbt wird. Diese Krankheitsform, die erst zu Beginn dieses Jahrhunderts beschrieben wurde, betrifft folglich vor allem Frauen.
Abzugrenzen ist ferner das erworbene Angioödem, das ebenfalls auf einem C1-INH-Mangel beruht, der jedoch durch einen erhöhten Abbau des Faktors verursacht ist. Ursache der Störung ist in der Regel eine Antikörperbildung gegen C1-INH. Es handelt sich damit um eine Autoimmunerkrankung. Sie entsteht meist im Rahmen einer lymphoproliferativen Erkrankung. Eine positive Familienanamnese besteht entsprechend nicht, genaue Häufigkeitsangaben sind bislang nicht möglich.
Diagnose meist erst nach Jahren
Akute Ödeme treten im Rahmen des HAE meist schon vor dem 20. Lebensjahr auf, allerdings dauert es derzeit im Durchschnitt rund 13 Jahre, ehe die richtige Diagnose gestellt wird. Denn die Erkrankung ist insgesamt betrachtet selten und folglich in der Öffentlichkeit kaum bekannt und auch im Bewusstsein der Ärzte nicht optimal verankert.
Hinzu kommt, dass die Patienten ganz unterschiedlich stark betroffen sind: So gibt es Menschen mit HAE, die zwölf und mehr Attacken pro Jahr erleiden. Rund ein Drittel der Betroffenen aber entwickelt zwischen sechs und elf Krankheitsphasen pro Jahr und ein weiteres Drittel leidet nur selten unter Symptomen. Ohne spezielle Behandlung halten die Schwellungen, die sich beim HAE nicht nur im Kopfbereich, sondern auch zum Beispiel im Bereich der Extremitäten und im Gastrointestinaltrakt manifestieren, meist zwei bis sogar fünf Tage an.
Hautödeme sind dabei die häufigste Manifestation der HAE. Die Schwellungen entwickeln sich zumeist allmählich über mehrere Stunden und damit deutlich langsamer als bei einem allergisch bedingten Angioödem. Ohne gezielte Behandlung verstärkt sich die Symptomatik im Normalfall über zwölf bis 36 Stunden und lässt dann in den folgenden Tagen langsam kontinuierlich wieder nach.
70 bis 80 Prozent der Patienten weisen neben den Hautödemen Magen-Darm- Attacken auf mit kolikartigen Schmerzen, Übelkeit, Erbrechen und Durchfall. Etwa jeder zweite Patient leidet davon abgesehen mindestens einmal in seinem Leben an einem Larynxödem, wobei sich die Schwellungen im Kehlkopfbereich innerhalb weniger Stunden zu einer kompletten Atemwegsobstruktion entwickeln können.
Fehldiagnosen und nicht indizierte invasive Eingriffe
Die Schwellung kann außerdem die Gehirnareale betreffen, die Blase, den Brustkorb, die Muskulatur, Gelenke, Nieren und die Speiseröhre. Kommt es zu Beschwerden, die auf eine Beteiligung dieser Organsysteme hinweisen, so wird an ein HAE üblicherweise nicht gedacht. Vor allem die unspezifischen gastrointestinalen Symptome führen häufig zu einer Fehldiagnose wie etwa der Diagnose einer Appendizitis oder eines Magengeschwürs mit zum Teil erheblichen Folgen für den Patienten. So gibt es Berichte, wonach bis zu einem Drittel der HAE-Patienten einem operativen Eingriff unterzogen werden, der aufgrund des nicht erkannten HAEs erfolgt, bei richtiger Diagnosestellung aber hätte verhindert werden können und der medizinisch nicht indiziert war.
Genaue Daten zur Prävalenz des hereditären Angioödems gibt es bislang nicht. Den Schätzungen zufolge ist weltweit etwa einer von 10 000 bis 50 0000 Menschen betroffen. Allerdings ist aufgrund der diagnostischen Probleme von einer hohen Dunkelziffer auszugehen.
In Deutschland wird die Zahl der Menschen mit einem hereditären Angioödem auf 1 200 geschätzt, in Europa soll sie bei 10 000 bis 50 000 liegen. Männer und Frauen erkranken in etwa gleich häufig, ethnische Unterschiede sind bei der Prävalenz nicht zu beobachten.
Auslöser sind meist unbekannt
Warum es im Rahmen des HAE immer wieder zu Attacken kommt und vor allem wie diese ausgelöst werden, bleibt im Einzelfall oft unerkannt. Allerdings gibt es verschiedene Faktoren, die offensichtlich die Ausbildung einer Attacke auslösen können: Dazu gehören Zahnextraktionen sowie operative Eingriffe wie eine Tonsillektomie. Offenbar können auch langes Sitzen oder Stehen sowie mechanische Belastungen, aber auch Hitze- und Kälteeinwirkungen sowie Chemikalien zur HAE-Attacke führen. Das gleiche gilt für emotionalen Stress, hormonelle Veränderungen, Infektionskrankheiten und auch einen Wetterumschwung.
Behandlung des HAE
Da das HAE nicht heilbar ist, besteht das Ziel der Behandlung primär darin, der durch Bradykinin gesteuerten Vasodilatation entgegenzuwirken und so die Schwellung zurückzubilden. Möglich ist dies zum Beispiel durch die Verabreichung von C1-INH-Konzentrat – eine Strategie, die darauf abzielt, den Inhibitormangel auszugleichen.
Die Substanz Icatibant, die eine ähnliche Struktur wie Bradykinin aufweist und dieses kompetitiv von den Bradykinin-B2-Rezeptoren des Gefäßendothels verdrängt, stellt ebenfalls einen effektiven Behandlungsansatz dar. Durch das Stoppen der Aktivierung des Rezeptors schließen sich die interzellulären Verbindungen wieder und es kommt zum Abklingen der Ödembildung und zum Rückgang der Schwellung. Icatibant hat sich bei Haut- wie auch bei Magen-Darm-Ödemen in Studien als wirksam und gut verträglich erwiesen. Der Bradykinin-Rezeptorantagonist kann als Fertigspritze subkutan verabreicht werden und bewirkt eine raschere Rückbildung des Ödems.
Christine VetterMerkenicher Str. 22450735 Köln


{kind=link}