Plattenepithelkarzinom bei einem Patienten mit Fanconi-Anämie
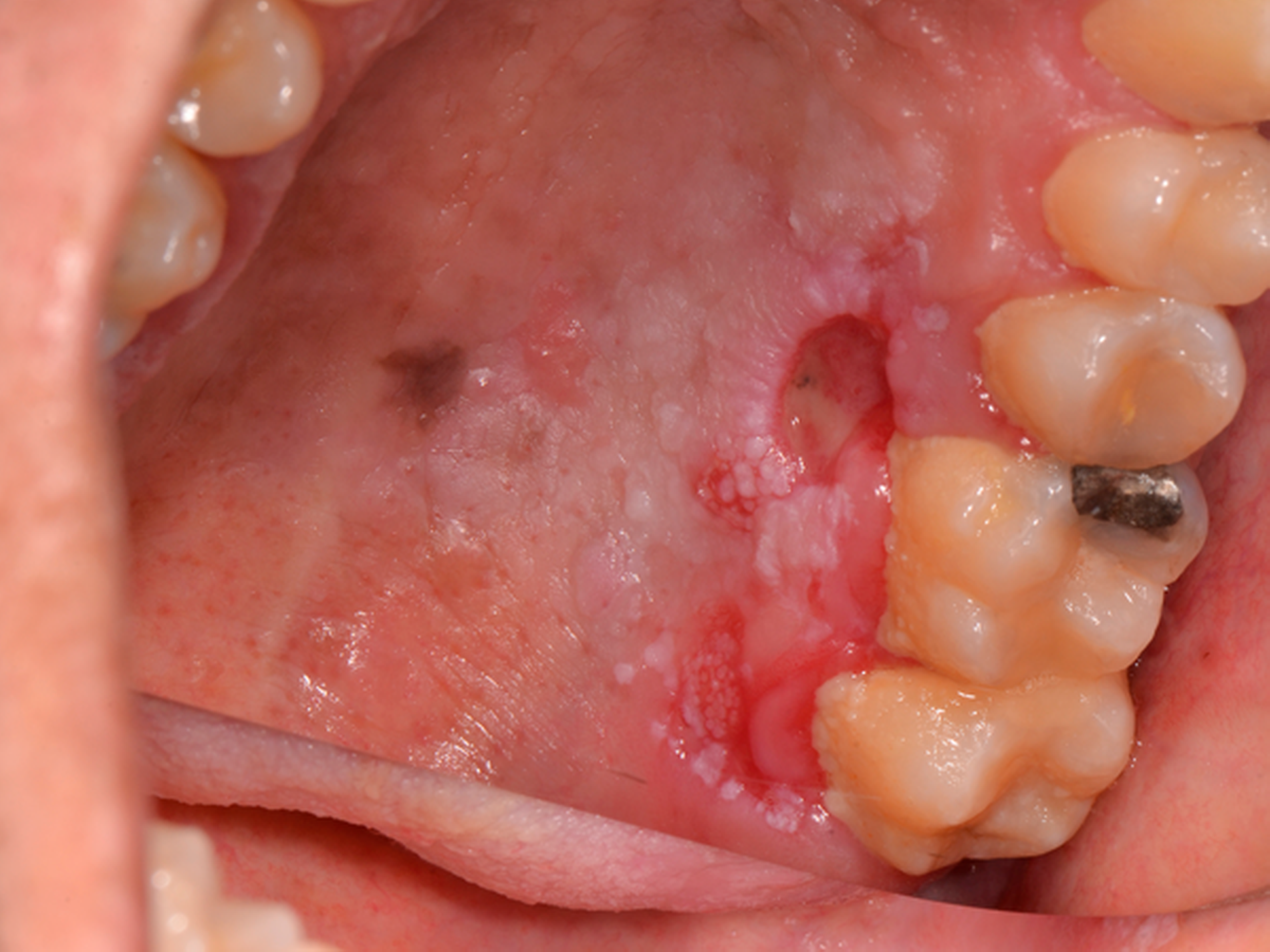
Die ambulante Erstvorstellung des Patienten erfolgte im Mai 2019 aufgrund einer neu aufgetretenen, über zwei Monate größenprogredienten Schleimhautveränderung im Bereich des Hartgaumens links (Abbildung 1). Die bereits auswärts durchgeführte Probeexzision ergab die Diagnose eines mäßig differenzierten Plattenepithelkarzinoms. Typische Risikofaktoren wie Alkohol- und Nikotinabusus lagen nicht vor. Als Grunderkrankung war jedoch eine Fanconi-Anämie bekannt. Im Alter von sechs Jahren war bei dem Patienten eine Knochenmarktransplantation durchgeführt worden.
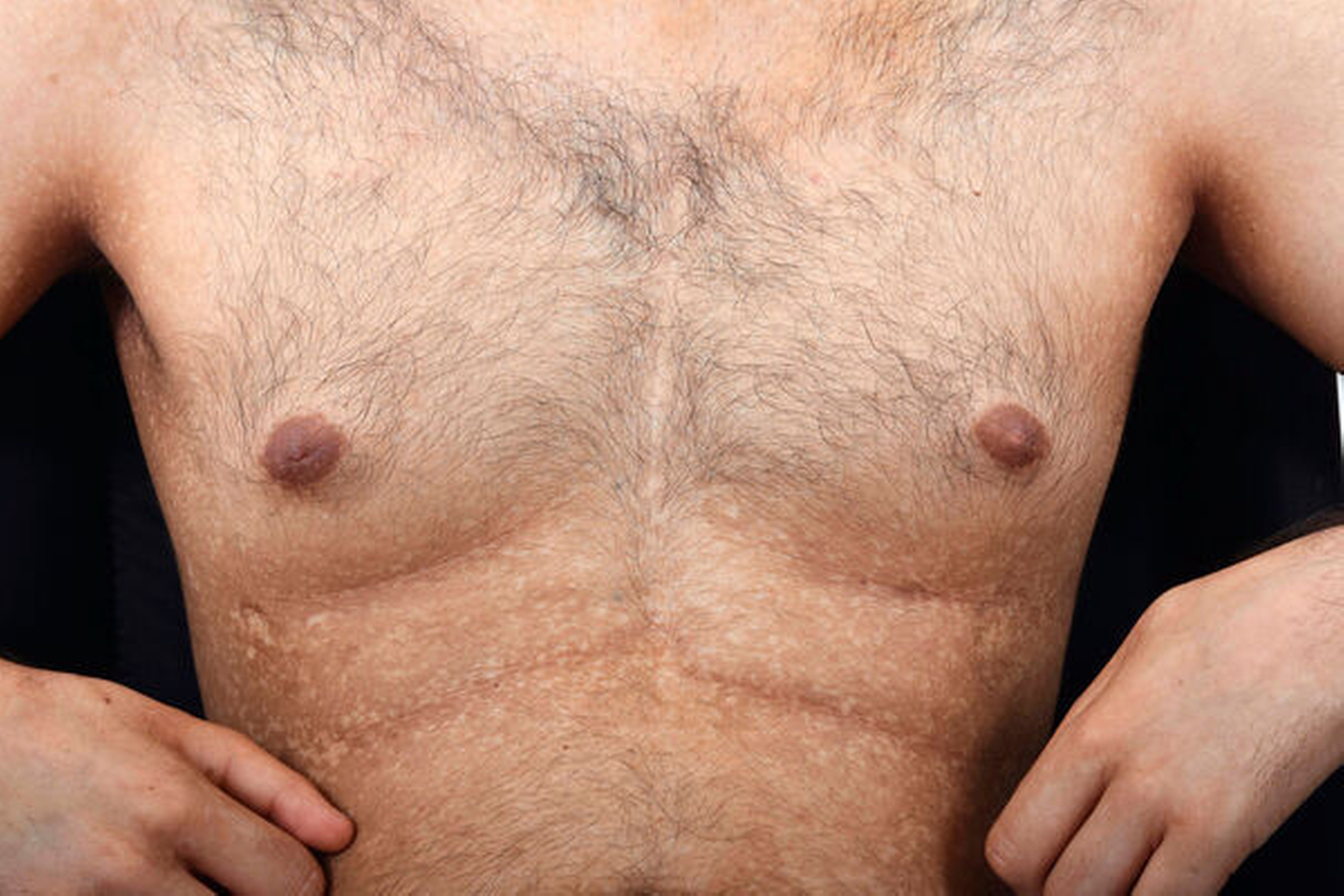
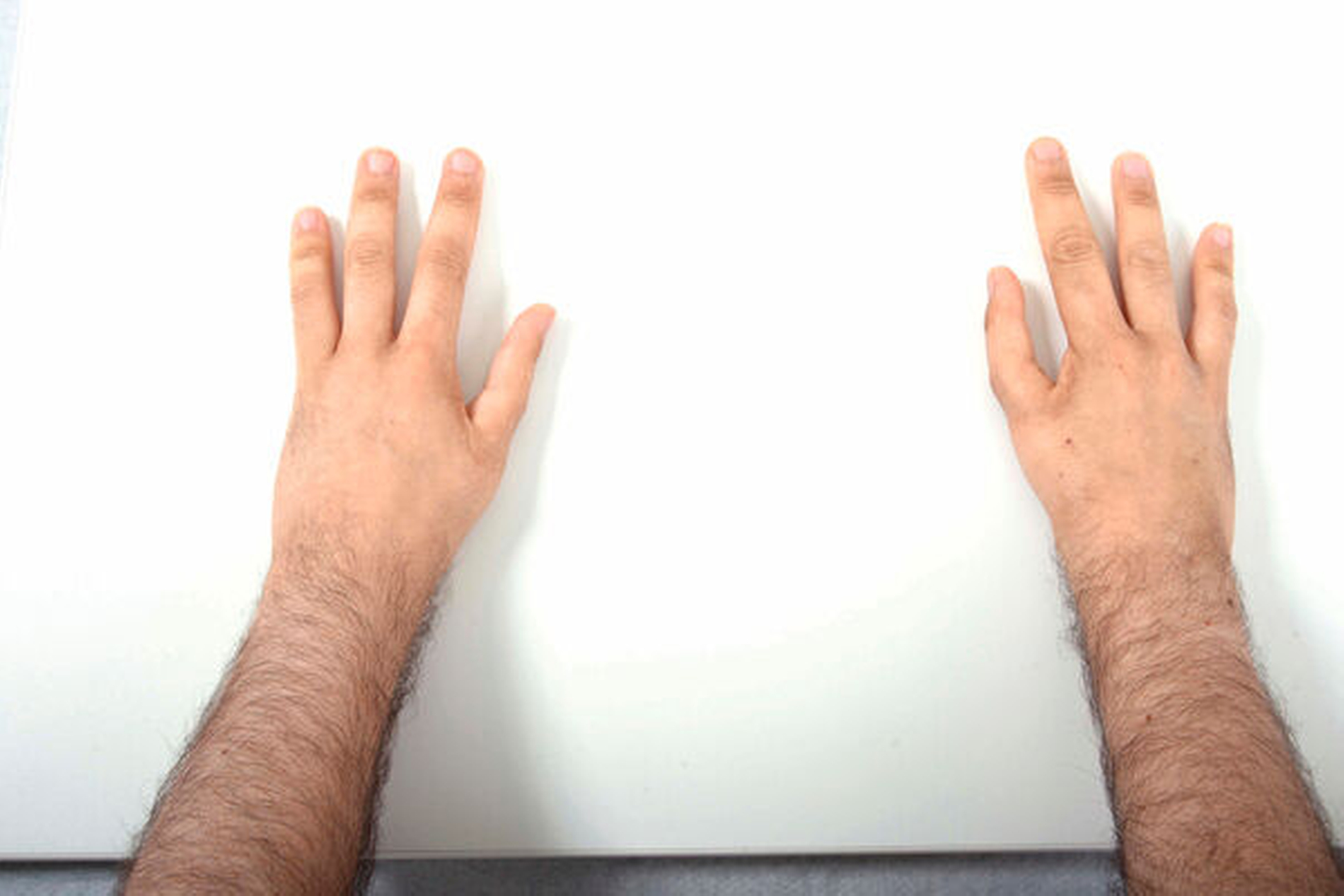
Klinisch präsentierte sich der Patient im minderwüchsigen Habitus und in gutem Allgemeinzustand. Bei Inspektion des restlichen Körpers fielen Café-au-Lait-Flecken im Bereich des Oberkörpers (Abbildung 2), eine Fehlbildung der Finger sowie beidseits iatrogen entfernte, kongenital-hypoplastische Daumen auf (Abbildung 3). Anamnestisch ließen sich zudem eine linksseitige Nierenagenesie und eine rechtsseitige Doppelniere, das beiderseitige Vorliegen einer Halsrippe und eine bei Geburt bestehende subvalvuläre Aortenstenose eruieren. Letztere war bereits operativ behandelt.
Nach der leitliniengerechten Tumorausbreitungsdiagnostik erfolgte die operative Therapie mittels Tumorresektion im Sinne einer Oberkieferteilresektion links, ipsilateraler selektiver Lymphknotenausräumung der Level I-III und eines weichgewebigen Defektverschlusses mittels mikrochirurgisch anastomosiertem faszio-kutanem Transplantat aus dem Bereich des antero-lateralen Oberschenkels (ALT). Auf eine knöcherne Rekonstruktion wurde in diesem Fall bewusst verzichtet, um den Eingriff auf das notwendige Minimum hinsichtlich Dauer, Ausdehnung und Wundflächen zu begrenzen. Hintergrund hierfür sind die weiter unten vorgestellten speziell zu berücksichtigenden Risiken bei der Behandlung von Patienten mit Fanconi-Anämie.
Die Operation verlief komplikationslos und der postoperative Verlauf gestaltete sich regelrecht. Der Patient befindet sich derzeit in engmaschigen Verlaufskontrollen und zeigte sich hier bislang mit klinisch unauffälligen Schleimhautverhältnissen ohne Anhalt für ein Rezidiv.
Diskussion
Die Fanconi-Anämie betrifft weniger als 1 von 300.000 Neugeborenen und stellt somit eine seltene genetische Erkrankung dar. Sie beruht auf einer Mutation in einem von derzeit 22 beschriebenen Fanconi-Anämie-Genen, die autosomal rezessiv vererbt wird oder – in sehr seltenen Fällen – spontan auftreten kann [Velleuer et al., 2020; Fiesco-Roa et al., 2019]. Die Folge ist eine in allen Zellen nachweisbare erhöhte Chromosomeninstabilität. Als Erstbeschreiber gilt der Schweizer Pädiater Guido Fanconi, der 1927 das Auftreten kongenitaler Defekte und einer Panzytopenie an drei Brüdern im Alter von fünf bis sieben Jahren beobachtete [Auerbach, 2009].
Hauptsymptom der Fanconi-Anämie ist die bei allen Patienten beobachtete fortschreitende Knochenmarkdepression mit Panzytopenie bis hin zur Aplastischen Anämie, die sich meist ab dem siebten Lebensjahr in voller Ausprägung zeigt. Betroffene leiden somit unter allen damit einhergehenden Symptomen wie rezidivierenden Infekten, erhöhter Blutungsneigung, Dyspnoe, verminderter Belastbarkeit sowie Entwicklungsstörungen. Etwa 70 Prozent der Patienten weisen zusätzliche kongenitale Fehlbildungen in unterschiedlicher Ausprägung auf. Beobachtet wurden vor allem Radiusstrahldysplasien, Hüftdysplasien, Skoliosen, hypoplastische oder aplastische Daumen, Mikrozephalie, Minderwuchs, Fehlbildungen innerer Organe und Azoospermie. Zudem haben Patienten häufig Pigmentstörungen in Form von Café-au-Lait-Flecken [Gillio et al., 1997; Auerbach, 2009].
Besonders gefährdet sind die betroffenen Patienten durch ein bereits in jungem Alter bestehendes, deutlich erhöhtes Krebsrisiko [Alter, 2003; Rosenberg et al., 2003; Niraj et al., 2019]. Etwa 60 Prozent erkranken dabei an einer Leukämie (insbesondere Akute myeloische Leukämie, Akute lymphatische Leukämie), an einem Myelodysplastischen Syndrom oder an soliden Tumoren, allen voran oralen Plattenepithelkarzinomen. Letztere äußern sich dann in einem besonders aggressiven Wachstumsverhalten mit erhöhten Rezidivraten und insgesamt schlechter Prognose [Kutler et al., 2016]. Die am häufigsten betroffene und in der Literatur beschriebene intraorale Lokalisation ist hierbei die Zunge [Millen et al., 1997], gefolgt von den restlichen Schleimhäuten in annähernd homogen verteilter Häufigkeit [Kutler et al., 2003].
Neben Plattenepithelkarzinomen im Mundhöhlen-, Oropharynx- und Larynxbereich finden sich zudem gehäuft anogenitale und dermale Plattenepithelkarzinome sowie Mammakarzinome. Nicht selten treten unterschiedliche Malignomentitäten auch synchron auf [Kutler et al., 2003].
Eine kausale Therapie der Grunderkrankung ist bislang nicht möglich, die Lebenserwartung der Patienten liegt bei circa 30 Jahren, kann jedoch durch die derzeit verfügbaren symptomatischen Therapiemöglichkeiten verlängert werden. Betroffene Patienten werden aktuell im Rahmen eines multimodalen Therapieansatzes in enger Anbindung an ein Spezialzentrum zunächst symptomatisch und supportiv mittels Transfusion von Zellbestandteilen, Verabreichung hämatopoetischer Wachstumsfaktoren, Hormontherapie und allogener Stammzelltransplantation behandelt. Letztere erhöht jedoch gleichzeitig das Risiko für das Auftreten solider Tumore deutlich und gilt somit als zusätzlicher Risikofaktor für das Auftreten von Plattenepithelkarzinomen [Millen et al., 1997; Rosenberg et al., 2003; Furquim et al., 2018].
Prinzipiell gelten für die Therapie von Plattenepithelkarzinomen im Kopf- und im Halsbereich in dieser Gruppe die gleichen Grundsätze wie bei allen anderen Patienten auch. Aufgrund der vorliegenden genetischen Konstitution sind hierbei allerdings wichtige Therapie- und Nachsorgeprinzipien zu berücksichtigen.
Einer möglichst frühzeitigen, regelmäßigen und engmaschigen Inspektion der Mundschleimhaut, die nach Anleitung zusätzlich in Eigenregie durchgeführt werden sollte, kommt hierbei eine besondere Bedeutung zu. So lassen sich suspekte Befunde bereits früh erkennen und eine zeitnahe Vorstellung in einer Mund-, Kiefer- und Gesichtschirurgischen Abteilung veranlassen. Das erstmalige Auftreten eines Plattenepithelkarzinoms wurde gehäuft bei jugendlichen Patienten im Alter von 15 Jahren beschrieben, vereinzelt auch bei wesentlich jüngeren [Kutler et al., 2016].
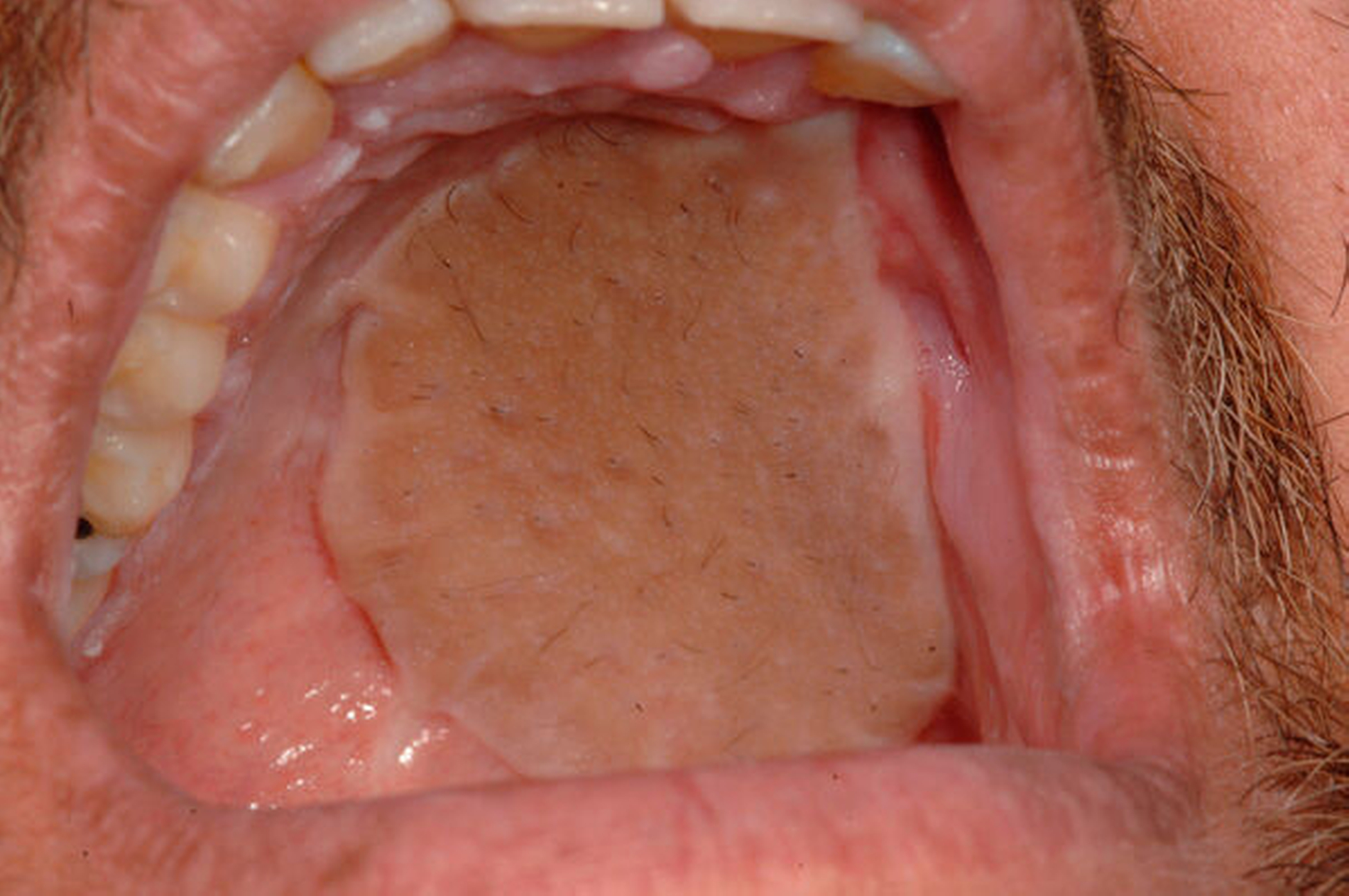
Auch bei Vorliegen kleinerer suspekter Läsionen sollte eine großzügige Indikation zur Probeexzision gestellt und einem abwartenden Vorgehen vorgezogen werden.
Nach Feststellung der Diagnose sollte eine leitliniengerechte, vorzugsweise operative Therapie mit kurativer Intention unter Berücksichtigung des deutlich erhöhten perioperativen Risikos und unter Einhaltung entsprechender Kautelen angestrebt werden [Wong et al., 2013; Kutler et al., 2016]. Diese hat auch deshalb einen hohen Stellenwert, da für betroffene Patienten aufgrund der vorliegenden multiplen genetischen Veränderungen eine interindividuell unterschiedlich ausgeprägte, im Allgemeinen verminderte Toleranz für adjuvante Therapien beschrieben wurde [Wong et al., 2013].
Die für eine Radiotherapie beobachteten Nebenwirkungen reichten – je nach angewandter Strahlendosis und individueller Strahlensensibilität – von Mukositis und Dysphagie bis hin zu ausgeprägter Zytopenie und Sepsis oder anderen beeinträchtigenden Nebenwirkungen, die nicht selten zu einem vorzeitigen Abbruch der Strahlentherapie führten [Alter, 2002; Birkeland et al., 2011; Kutler et al., 2016]. Mitunter letale Nebenwirkungen wurden auch für die Anwendung unterschiedlicher Chemotherapeutika berichtet, so zum Beispiel eine Myelodepression unter Cisplatin [Birkeland et al., 2011; Wong et al., 2013; Kutler et al., 2016]. Sollte die Anwendung (neo-)adjuvanter Therapieverfahren im Fall eines fortgeschrittenen Stadiums indiziert sein, ist dies nur nach genauer Abwägung aller Risiken und unter Einbeziehung aller beteiligten Fachdisziplinen in Erwägung zu ziehen.
Die größte Bedeutung kommt auch postoperativ den engmaschigen Verlaufskontrollen zu. Die Intervalle sind hierbei enger als im Normalfall zu halten, da bei Patienten mit einer Fanconi-Anämie von wesentlich früher auftretenden lokoregionären Rezidiven berichtet wurde. Häufig wurden Intervalle von vier bis acht Wochen diskutiert [Furquim et al., 2018].
Da es derzeit keine allgemeingültigen Richtlinien für die Therapie von Plattenepithelkarzinomen bei Fanconi-Anämie-Patienten gibt, sollte eine sinnvolle Abwägung für jeden einzelnen Patienten unter Berücksichtigung des Alters, des Tumorstadiums sowie des Ausprägungsgrades der genetischen Mutation und der damit einhergehenden Gesamtkonstellation des Patienten getroffen werden. Einigkeit besteht jedoch darüber, dass alle Patienten von engmaschigen klinischen Verlaufskontrollen und einer frühzeitigen operativen Herangehensweise profitieren. Diese sollte sich dabei auf das notwendige Maß begrenzen, um die Operationsdauer, die Morbidität und somit die perioperativen Risiken weitestgehend zu reduzieren. Im hier beschriebenen Fall wurde daher bewusst auf eine primäre knöcherne Rekonstruktion verzichtet.
Sameena Sandhu
Klinik und Poliklinik für Mund-, Kiefer- und Gesichtschirurgie,
Universitätsklinikum Heidelberg
Im Neuenheimer Feld 400, 69120 Heidelberg
sameena.sandhu@med.uni-heidelberg.de
Univ.-Prof. Dr. Dr. Dr. h.c. Jürgen Hoffmann
Klinik und Poliklinik für Mund-, Kiefer- und Gesichtschirurgie,
Universitätsklinikum Heidelberg
Im Neuenheimer Feld 400, 69120 Heidelberg
PD Dr. Dr. Oliver Ristow
Klinik und Poliklinik für Mund-, Kiefer- und Gesichtschirurgie,
Universitätsklinikum Heidelberg
Im Neuenheimer Feld 400, 69120 Heidelberg
Fazit für die Praxis
Bei jungen Patienten mit suspekten Schleimhautbefunden ohne Vorliegen typischer Risikofaktoren können auch seltene Ursachen – wie eine Fanconi-Anämie – vorliegen. Sollte diese Diagnose aufgrund der ausgeprägten phänotypischen Heterogenität noch nicht im Raum stehen, ist eine dahingehende genetische Abklärung zu empfehlen.
Aufgrund des aggressiven Wachstumsverhaltens sind eine zeitnahe operative Therapie und sehr engmaschige Verlaufskontrollen angeraten.
Die ausgeprägten perioperativen Risiken machen eine patientenspezifische Therapie in einem interdisziplinären Setting notwendig.
Literaturliste
Alter, B. P. (2002). „Radiosensitivity in Fanconi's anemia patients.“ Radiother Oncol 62(3): 345-347.
Alter, B. P. (2003). „Cancer in Fanconi anemia, 1927–2001.“ Cancer 97(2): 425-440.
Auerbach, A. D. (2009). „Fanconi anemia and its diagnosis.“ Mutat Res 668(1-2): 4-10.
Birkeland, A. C., et al. (2011). „Postoperative clinical radiosensitivity in patients with fanconi anemia and head and neck squamous cell carcinoma.“ Archives of Otolaryngology–Head & Neck Surgery 137(9): 930-934.
Fiesco-Roa, M. O., et al. (2019). „Genotype-phenotype associations in Fanconi anemia: a literature review.“ Blood reviews 37: 100589.
Furquim, C. P., et al. (2018). „Oral cancer in Fanconi anemia: Review of 121 cases.“ Crit Rev Oncol Hematol 125: 35-40.
Gillio, A. P., et al. (1997). „Phenotypic consequences of mutations in the Fanconi anemia FAC gene: an International Fanconi Anemia Registry study.“ Blood, The Journal of the American Society of Hematology 90(1): 105-110.
Kutler, D. I., et al. (2003). „High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia.“ Archives of Otolaryngology–Head & Neck Surgery 129(1): 106-112.
Kutler, D. I., et al. (2016). „Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up.“ Laryngoscope 126(4): 870-879.
Millen, F., et al. (1997). „Oral squamous cell carcinoma after allogeneic bone marrow transplantation for Fanconi anaemia.“ British journal of haematology 99(2): 410-414.
Niraj, J., et al. (2019). „The Fanconi anemia pathway in cancer.“ Annual review of cancer biology 3: 457-478.
Rosenberg, P. S., et al. (2003). „Cancer incidence in persons with Fanconi anemia.“ Blood 101(3): 822-826.
Wong, W. M., et al. (2013). „Squamous cell carcinoma of the oral tongue in a patient with Fanconi anemia treated with radiotherapy and concurrent cetuximab: A case report and review of the literature.“ Head & neck 35(10): E292-E298.
Velleuer, E., Dietrich, R., Pomjanski, N., de Santana Almeida Araujo, I.K., Silva de Araujo, B.E., Sroka, I., Biesterfeld, S., Böcking, A. and Schramm, M. (2020), Diagnostic accuracy of brush biopsy–based cytology for the early detection of oral cancer and precursors in Fanconi anemia. Cancer Cytopathology, 128: 403-413. doi.org/10.1002/cncy.22249

Sameena Sandhu
Im Neuenheimer Feld 400, 69120 Heidelberg

Prof. Dr. Dr. Jürgen Hoffmann
ZMK-Heilkunde Klinik und Poliklinik für MKG-Chirurgie
Im Neuenheimer Feld 400
69120 Heidelberg

Dr. Oliver Ristow
Klinik und Poliklinik für\r\n
Mund-, Kiefer- und\r\n
Gesichts chirurgie, Univer -\r\n
sitätsklinikum Heidelberg\r\n
Im Neuenheimer Feld 400\r\n
69120 Heidelberg\r\n
")
