Maligner peripherer Nervenscheidentumor

Ein 73-jähriger Mann wurde wegen eines am Kinn befindlichen Tumors mit der Bitte um Übernahme der weiteren Diagnostik und Therapie durch die Kollegen der Klinik und Poliklinik für Dermatologie im Hause konsiliarisch vorstellig. Der Patient berichtete, der Befund bestehe seit mehreren Monaten und sei ihm erstmals durch eine Schnittverletzung bei der morgendlichen Rasur aufgefallen. Bei der klinischen Inspektion imponierte mental links ein etwa 1,5 bis 2 cm großer, derb-knotiger Tumor mit Ulzeration der Haut (Abbildung 1). Die Mundschleimhaut war unauffällig. Sensibilitätsstörungen oder Schmerzen lagen nicht vor. Vom Patienten wurde lediglich eine geringgradige Missempfindung im Bereich des Tumors angegeben, die er als „leichtes Ziehen“ beschrieb. Durch die zuweisenden Kollegen war unter der Verdachtsdiagnose einer epidermalen Zyste zunächst eine bioptische Befundsicherung vorgenommen worden. Nach histopathologischer Aufarbeitung ergab sich jedoch ein mittelgradig differenziertes neurogenes Malignom, entsprechend einem malignen peripheren Nervenscheidentumor.
In einer im Rahmen des umgehend eingeleiteten Tumorstagings angefertigten Computertomographie sowie einer Magnetresonanztomographie des Halses und des Unterkiefers zeigte sich ein etwa 2 cm großer, gering Kontrastmittel aufnehmender, weichteildichter Tumor ventral des Unterkiefers mit Infiltration der mimischen Muskulatur. Der Befund reichte bis an das mandibuläre Periost heran, jedoch gab es keine Anzeichen einer knöchernen Destruktion (Abbildungen 2 und 3). Submental rechts, unmittelbar caudal des Tumors, fand sich ein auf etwa 1,5 cm vergrößerter, metastasensuspekter Lymphknoten. Weitere suspekte Lymphknoten lagen submandibulär links. Die Positronenemissionstomographie ergab einen erhöhten Glukosemetabolismus im Bereich des Tumors und des benachbarten Lymphknotens. Fernmetastasen konnten nicht nachgewiesen werden.
Nach Abschluss der Diagnostik erfolgte die Resektion des Tumors mit großem Sicherheitsabstand (Abbildung 4) unter Mitnahme der Außenkortikalis des Unterkiefers und supraomohyoidaler Lymphknotenausräumung beidseits. Der Resektionsdefekt wurde mittels eines mikrovaskulär anastomosierten Latissimus-dorsi-Lappens gedeckt.
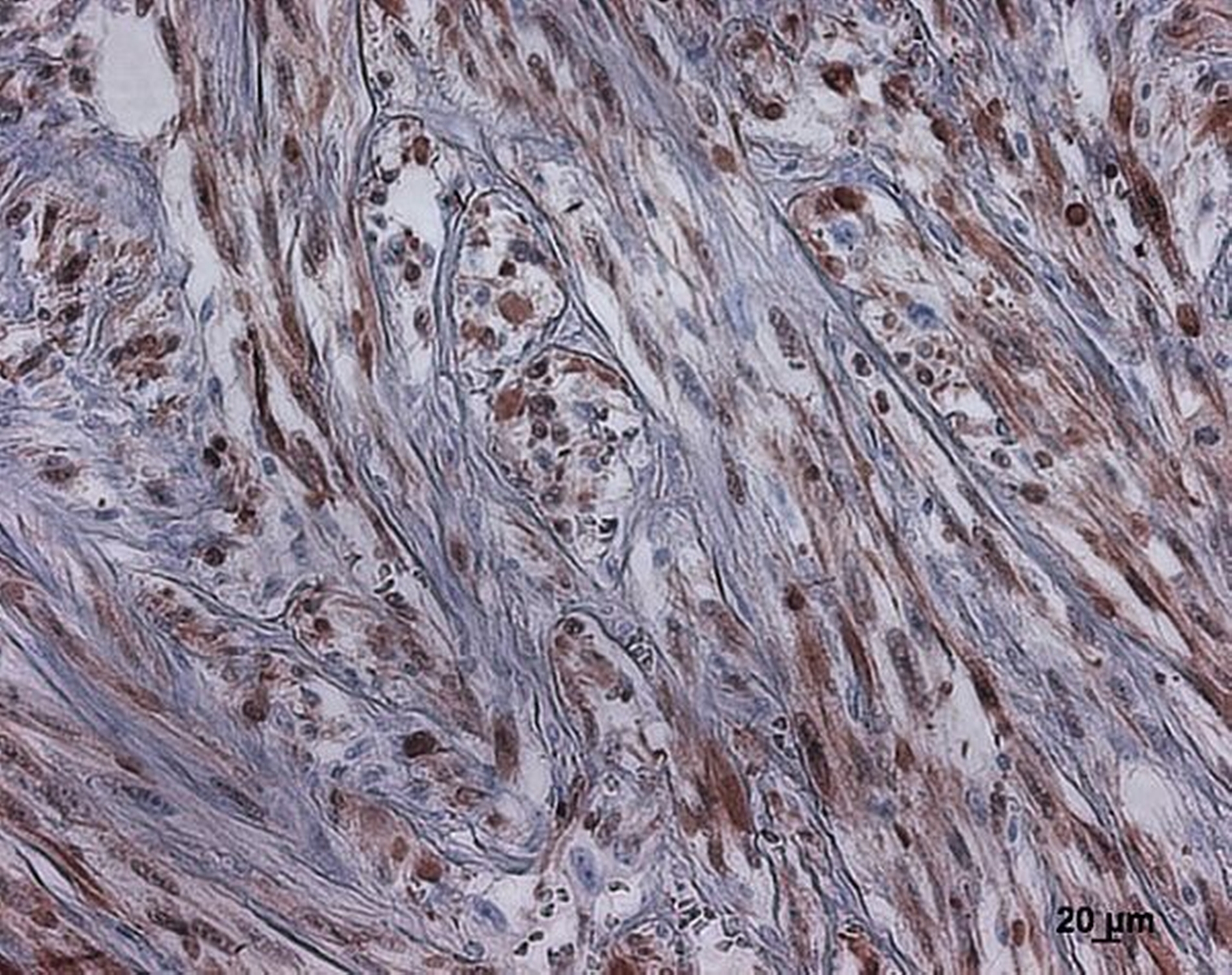
Im endgültigen histopathologischen Gutachten ergab sich nun ein geringgradig differenzierter mesenchymaler Tumor. (Abbildungen 5, 6 und 7). Immunhistochemisch wurde S-100 nachgewiesen (Abbildung 8). In der Zusammenschau dieser Befunde wurde der Tumor als maligner peripherer Nervenscheidentumor eingestuft. Der resezierte Unterkieferknochen war nicht infiltriert. Die Resektion erfolgte lokal in sano. Lymphknotenmetastasen konnten nicht nachgewiesen werden. Es ergab sich folgende abschließende TNM-Klassifikation: G 3, pT 1a, pN 0 (0/19), L 0, V 0, R 0.
Postoperativ wurde der Patient entsprechend den Empfehlungen der Literatur einer adjuvanten Radiotherapie zugeführt.
Diskussion
Malignome, die von peripheren Nerven ausgehen oder eine Nervenscheidendifferenzierung erkennen lassen, werden definitionsgemäß als maligne periphere Nervenscheidentumoren (Malignant peripheral nerve sheath tumors, MPNST) eingestuft. Die in der Vergangenheit benutzten Synonyme wie Neurofibrosarkom, malignes Schwannom und neurogenes Sarkom sollten nicht mehr verwendet werden, da sich die Tumoren nur äußerst selten aus den primär benignen Schwannomen entwickeln und auch keine fibroblastischen oder mesodermalen Anteile enthalten [Wong et al., 1998; Minovi et al., 2007].
Die Mehrzahl der MPNST entsteht zu etwa gleichen Teilen sporadisch oder entwickelt sich aus Neurofibromen. Sie sind daher häufig mit einer Neurofibromatose, insbesondere der Neurofibromatose Typ-1 (M. Recklinghausen) vergesellschaftet, weshalb diese als prädisponierend angesehen wird [Minovi et al., 2007; Anghileri et al., 2006; Ducatman et al., 1986]. Eine vorangegangene Radiotherapie aus anderer Indikation gilt ebenfalls als Risikofaktor. [Ducatman et al., 1983]. Die jährliche Inzidenz maligner peripherer Nervenscheidentumoren in der Gesamtpopulation wird mit 1/100 000 angegeben. Die Tumoren sind damit im allgemeinen Patientenkollektiv selten, sie machen jedoch insgesamt etwa fünf Prozent aller Weichgewebssarkome aus [Wong et al., 1998; Minovi et al., 2007]. Das Hauptmanifestationsalter liegt zwischen der dritten und der fünften Lebensdekade, wobei die Verteilung zwischen den Geschlechtern etwa gleichmäßig ist.
MPNST finden sich bevorzugt am Rumpf und an den Extremitäten, das Auftreten an Kopf und Hals ist mit etwa 10 bis 20 Prozent eher selten [Minovi et al., 2007].
Therapeutisch wird die Resektion mit großem Sicherheitsabstand, kombiniert mit einer adjuvanten Radiotherapie empfohlen [Wong et al., 1998; Minovi et al., 2007].
Die MPNST sind sehr aggressive Tumoren mit hoher Lokalrezidivrate (40 bis 65 Prozent) und hoher Fernmetastasierungsrate (16 bis 68 Prozent). Die Filialisierung erfolgt überwiegend hämatogen, weshalb die Ausräumung der Lymphabflussgebiete nicht als obligatorisch angesehen wird und Fällen mit klinisch oder radiologisch suspekten Lymphknoten wie bei unserem Patienten vorbehalten bleibt [Minovi et al., 2007]. Metastasen finden sich häufig in der Lunge, gefolgt von Leber und Lymphknoten, seltener in Knochen, Weichgewebe, Nieren, Nebennieren, Hirn oder Ovarien [Ducatman et al., 1986].
Die Prognose aller malignen peripheren Nervenscheidentumoren ist trotz radikaler Therapie mit einer Fünf-Jahres-Überlebensrate von etwa 50 Prozent schlecht. Bei Manifestationen an Kopf und Hals ist die Prognose wegen der anatomischen Verhältnisse und der damit häufig verbundenen Schwierigkeit der vollständigen Resektion mit ausreichendem Abstand noch schlechter. Hier werden Fünf-Jahres-Überlebensraten von nur 15 bis 20 Prozent angegeben [Minovi et al., 2007].
In der Histopathologie zeigen MPNST in der Regel einen faszikulären Aufbau aus spindeligen Tumorzellen mit teils buckeligen, teils längsovalen Zellkernen. Der Wechsel von zelldichten Arealen und hypozellulären, myxoiden Zonen erzeugt einen marmorierten Aspekt des Tumors. Selten finden sich eine nukleäre Palisadierung und hyaline Banden. Immunhistochemisch wird neben unspezifischen Markern wie Leu-7 und PGP9.5 am häufigsten der zuverlässige Marker S-100 verwendet. 50 bis 90 Prozent aller MPNST sind – wie auch im hier vorgestellten Fall – fokal S-100 positiv. Um MPNST von Neurofibromen abzugrenzen, kann eine p53-Färbung ergänzt werden. Die MPNST zeigen in 50 Prozent der Fälle eine positive Immunreaktion für p53 [Weiss et Goldblum, 2001].
Differentialdiagnostisch müssen andere Malignomentitäten, aber auch benigne Weichgewebstumoren in Betracht gezogen werden. Mittels Computertomographie und Magnetresonanztomographie kann häufig bereits im Vorfeld zwischen benignen und malignen Tumoren differenziert werden [Hems et al., 1997]. Auch das klinische Bild kann Hinweise geben, da mit malignen peripheren Nervenscheidentumoren im Gegensatz zu benignen Tumoren nicht selten neurologische Ausfälle einhergehen.
Die Diagnose wird schließlich histopathologisch gestellt. In den meisten Fällen fällt es leicht, die MPNST als Malignome zu diagnostizieren. Um benigne Tumoren mit neurogener Differenzierung wie das Neurofibrom oder das Schwannom histopathologisch abzugrenzen, spielen eindeutige Malignitätskriterien wie eine erhöhte Zellularität des Tumors, zelluläre Atypien und eine erhöhte mitotische Aktivität eine Rolle [Weiss et Goldblum, 2001]. Die Differenzierung von anderen Sarkomen, wie Fibrosarkom, Synovialsarkom oder Leiomyosarkom, stellt die eigentliche Herausforderung dar. Morphologisch zeigt das Fibrosarkom ein symmetrisches, uniformes Wachstumsmuster, während das Synovialsarkom epithelial differenzierte Abschnitte aufweist. Hier können immunhistochemische Färbungen bei der Diagnosestellung helfen. Während MPNST für Zytokeratin immer negativ sind, zeigt das Synovialsarkom hierfür meist eine positive Färbereaktion. Leiomyosarkome können in der Regel rein histomorphologisch eindeutig von MPNST abgegrenzt werden [Weiss et Goldblum, 2001].
Dr. Stefan LegalPD Dr. Dr. Ulrich WahlmannDr. Dr. Martin GosauKlinik und Poliklinik für Mund-, Kiefer- und GesichtschirurgieUniversität RegensburgFranz-Josef-Strauß-Allee 1193053 Regensburg
Katharina ZeitlerInstitut für PathologieUniversität RegensburgFranz-Josef-Strauß-Allee 1193053 Regensburg

