Kleiner Enzymmangel mit großen Folgen
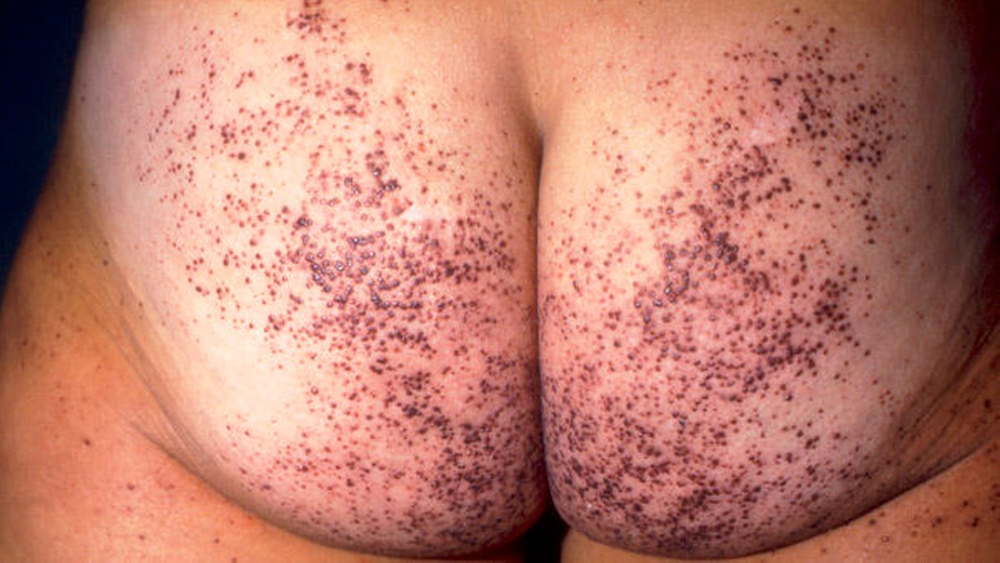
- Typisches Angiokeratom beim Morbus Fabry © Shire


Obwohl sie zu den seltenen Erkrankungen zählen, unterscheidet man bei den lysosomalen Speicherkrankheiten rund 45 verschiedene Krankheitsbilder. Es handelt sich um genetisch bedingte Stoffwechselerkrankungen mit progredientem Verlauf, die sich an unterschiedlichen Organsystemen manifestieren können und oft eine große phänotypische Variabilität aufweisen – was erklärt, warum die jeweiligen Erkrankungen oft erst nach zum Teil mehrfachen Fehldiagnosen und mit einer jahrelangen Verzögerung richtig diagnostiziert werden.
Die einzelnen Krankheitsbilder beruhen auf einem genetisch bedingten Enzymdefekt, der zur Folge hat, dass der Abbau von Stoffwechselendprodukten in den Lysosomen gestört ist und die Abbauprodukte nicht adäquat entsorgt werden können. Dadurch kommt es zur pathologischen Akkumulation in den Zellen, was massive Schädigungen der betroffenen Organsysteme zur Folge haben kann.
Quellen und weitere Informationen:
Gesellschaft für Mukopolysaccharidosen e. V., www.mps-ev.de
Lysosomale Speicherkrankheiten, www.lysosolutions.de
Morbus Fabry, www.fabry-im-fokus.de
Morbus Gaucher, www.ggd-ev.de
Der Gendefekt kann verschiedene lysosomale Enzyme betreffen, was die Vielzahl der Erkrankungen erklärt. Das betreffende Enzym kann in seiner Funktion von Patient zu Patient unterschiedlich stark beeinträchtigt sein, so dass es erhebliche Unterschiede im Krankheitsverlauf gibt, wodurch die Diagnostik zusätzlich erschwert wird. Abhängig vom vorliegenden Enzymdefekt gibt es die Mukopolysaccharidosen, die Glykoproteinosen, die Sphingolipidosen und die neuronalen Zeroidlipofuszinosen.
Die bekanntesten lysosomalen Speicherkrankheiten sind der Morbus Hunter, der zu den Mukopolysaccharidosen zählt, der Morbus Fabry und der Morbus Gaucher, die den Sphingolipidosen zuzuordnen sind, sowie der Morbus Pompe, der auf einem Defekt der lysosomalen Alpha-Glucosidase beruht.
Morbus Gaucher
Der Morbus Gaucher stellt die häufigste Speicherkrankheit dieser Art dar. Sie basiert auf einem Mangel an ß-Glukozerebrosidase, einem Enzym des Sphingolipidstoffwechsels. Infolgedessen reichert sich das Abbauprodukt Glukozerebrosid insbesondere in den Makrophagen an, weil die gespaltenen Glykolipide vor allem beim Abbau von Lymphozyten und Erythrozyten in Makrophagen anfallen. Mit zunehmender Speicherungschwellen die Makrophagen zu sogenannten Gaucher-Zellen an. Die Gaucher-Speicherzellen sammeln sich in verschiedenen Organen, etwa in der Leber, in der Milz und im Knochenmark. Das erklärt die charakteristischen Symptome der Erkrankung wie die Hepatosplenomegalie in Verbindung mit einer Anämie, einer Thrombozytopenie sowie einer erhöhten Blutungsneigung. Die Patienten klagen in der Regel über eine rasche Ermüdbarkeit, Leistungsschwäche, Konzentrationsschwierigkeiten und eine erhöhte Infektanfälligkeit.
Im Verlauf der Erkrankung können außerdem gravierende Komplikationen im Bereich der Knochen auftreten bis hin zum Knocheninfarkt, schmerzhaften Knochenkrisen und Frakturen. Viele Patienten entwickeln allerdings lediglich viszerale Symptome. Möglich sind auch neuropathische Krankheitsverläufe.
Seit Anfang der Neunzigerjahre gibt es mit der Enzymersatztherapie beim Morbus Gaucher eine spezifische Behandlungsmöglichkeit. Bei eher milder Symptomatik ist außerdem eine Substratreduktionstherapie möglich.
Morbus Fabry
Der Morbus Fabry beruht auf einem X-chromosomal vererbten Mangel des lysosomalen Enzyms α-Galaktosidase A. Hierdurch kommt es zu Funktionsschädigungen an verschiedenen Organen und Geweben. Die Symptome sind vielgestaltig und treten nicht nur bei Erwachsenen, sondern oft bereits ab dem zweiten Lebensjahr auf, werden aber häufig nicht richtig erkannt. Vor allem durch Morbus Fabry bedingte Schmerzen bei Kindern werden oftmals als „Wachstumsschmerz“ fehlgedeutet.
Betroffen sind vor allem die Haut, die Augen, das Herz und die Nieren sowie das periphere und das zentrale Nervensystem. Geklagt wird typischerweise über Schmerzen, eine Hitzeintoleranz, gastrointestinale Beschwerden, Parästhesien in Fingern und/oder Zehen, ein Hörverlust und Augenprobleme. Häufig kommt es zu Hautläsionen, zu einer kardialen und/oder renalen Dysfunktion, nicht selten auch zum Auftreten eines Schlaganfalls vor dem 55. Lebensjahr.
Allerdings zeigt die Mehrzahl der Patienten nicht das Vollbild der Erkrankung, was die Diagnosestellung erschwert. Es dauert Schätzungen zufolge rund zwölf Jahre vom ersten Symptom bis zur richtigen Diagnose. In der Regel verstärken sich die Symptomatik wie auch die Organschäden im Verlauf der Erkrankung, was die Bedeutung einer frühzeitigen Diagnose unterstreicht.
Der Morbus Fabry beeinträchtigt nicht nur die Lebensqualität, sondern reduziert auch die Lebenserwartung. Sie ist bei Frauen um circa 15 Jahre, bei Männern um schätzungsweise 20 Jahre verkürzt. Die häufigsten Todesursachen sind ein Nierenversagen, eine Kardiomyopathie sowie zerebrale Ereignisse wie beispielsweise ein Schlaganfall. Mittlerweile kann die Erkrankung ebenso wie der Morbus Gaucher durch eine Enzymersatztherapie behandelt werden.
Morbus Pompe
Dem Morbus Pompe liegt ein autosomal-rezessiv vererbter Mangel des Enzyms α-1,4-Glukosidase zugrunde, was eine Akkumulation von Glykogen in den Lysosomen und im weiteren Verlauf in den Zellen selbst zur Folge hat. Fehlt das Enzym komplett, entwickelt sich bereits in den ersten Lebensmonaten eine Kardiomyopathie – und die Kinder versterben unbehandelt schon im ersten Lebensjahr. Sie fallen oft früh durch eine Muskel- sowie eine Trinkschwäche auf, strengen sich zum Beispiel beim Trinken übermäßig an, schwitzen auffällig und sind oft rasch erschöpft. Auffällig ist auch eine verzögerte Gewichtsentwicklung. Im Vergleich mit Gleichaltrigen ist der Muskeltonus gering: Die Kinder sind oft unfähig, den Kopf selbstständig anzuheben, zeigen kaum Krabbelaktivität und eine insgesamt reduzierte Spontanbewegung.
Liegt noch eine Restaktivität des Enzyms vor, ist der Krankheitsverlauf milder und die Erkrankung manifestiert sich oft erst im Jugend- oder sogar erst im Erwachsenenalter. Der Enzymdefekt wirkt sich dabei insbesondere in der Herz- und in der Skelettmuskulatur aus und es kommt zu einer progredienten Myopathie. Neben einer fortschreitenden Schwäche der Bewegungsmuskulatur resultieren oft deutliche Atemprobleme. Auffällig sind zudem eine Erhöhung der Kreatininkinase sowie der Transaminasen (GOT und GPT). Im Verdachtsfall lässt sich die Diagnose durch eine Enzymbestimmung sichern.
Aus Sicht der Zahnmedizin: Lyosomale Speicherkrankheiten
Die lysosomalen Speicherkrankheiten zählen zu den seltenen Erkrankungen mit teilweise typischen Veränderungen im orofazialen Bereich. Für einige gibt es bereits Enzymersatztherapien, die bei symptomatischen Patienten eingesetzt werden, die Symptomlast senken und die Lebensqualität der Patienten verbessern.
Der Morbus Gaucher ist mit einer Inzidenz von 1:40.000 die häufigste der rund 50 Speicherkrankheiten. Typische Symptome sind Hepatosplenomegalie, Anämie, Thrombozytopenie und Skelettbeteiligung. Orofaziale Manifestationen sind seltener und in der Regel asymptomatisch, können jedoch bei der zahnmedizinischen Routinediagnostik festgestellt werden. Die Infiltration der Mandibel durch Gaucher-Zellen ist neben der Infiltration der langen Röhrenknochen ein typisches Symptom. Die Maxilla ist hingegen aufgrund der anderen Knochenstruktur deutlich seltener betroffen. Radiologisch zeigen sich pseudozystische oder honigwabenartige Läsionen vor allem in der Prämolaren-Molaren-Region. Eine verstärkte Osteopenie und ein Verlust der trabekulären Architektur in den Läsionen sind ebenfalls typisch. Eine Biopsie der Läsion ist weder notwendig noch empfohlen. Vielmehr sollte durch ein Enzym-Assay die Diagnose gestellt werden.
Die Läsionen sind keine Indikation für die Extraktion der benachbarten Zähne, auch Implantationen sind nicht kontraindiziert. Bei zahnärztlich chirurgischen Eingriffen ist jedoch zu berücksichtigen, dass ein erhöhtes Blutungsrisiko besteht, das allerdings mit lokalen Maßnahmen in der Regel beherrscht werden kann.
Assoziiert mit lysosomalen Speichkrankheiten können auch Syndrome diagnostiziert werden, bei denen es zu Gingivahyperplasien, Zahnfehlbildungen, Makroglossie und mehr kommt. Wichtig bei der häufig lebenslangen Betreuung dieser Patienten sind die gute interdisziplinäre Zusammenarbeit der involvierten Disziplinen und die Therapie in spezialisierten Zentren, um eine adäquate Versorgung zu gewährleisten.
Bei Orphanet, dem Referenz-Portal für Informationen über seltene Krankheiten und Orphan Drugs, gibt es als umfassende Informationen:
ein Verzeichnis und eine Klassifikation der seltenen Krankheiten,
die Orphanet-Enzyklopädie,
das Leistungsverzeichnis: Expertenzentren, Diagnostikleistungen, Forschungsprojekte, Register, klinische Studien, Patientenorganisationen,
eine Liste der Orphan Drugs sowie
Leitlinien, Berichte.
Univ.-Prof. Dr. Dr. Monika Daubländer
Leitende Oberärztin der Poliklinik für Zahnärztliche Chirurgie
Universitätsmedizin der Johannes Gutenberg-Universität Mainz
Poliklinik für Zahnärztliche Chirurgie
Augustusplatz 2, 55131 Mainz
daublaen@uni-mainz.de
PD Dr. Dr. Peer W. Kämmerer
Klinik und Poliklinik für Mund-, Kiefer- und Plastische Gesichtschirurgie der Universität Rostock
Schillingallee 35, 18057 Rostock
Inzwischen ist für alle Verlaufsformen des Morbus Pompe eine Enzymersatztherapie verfügbar. Das fehlende Enzym α-Glukosidase wird dabei alle zwei Wochen als Infusion verabreicht, wodurch sich die Prognose – insbesondere der betroffenen Säuglinge – erheblich verbessert.
Morbus Hunter
Der Morbus Hunter ist einfacher zu diagnostizieren, weil die Patienten deutlich sichtbare Merkmale zeigen wie vergröberte Gesichtszüge mit abgeflachter Nase, vorgewölbter Stirn, vollen Lippen, einer vergrößerten Zunge, einer Prognathie, verdicktem, buschigem Haar sowie buschigen Augenbrauen bei meist gleichzeitiger Makrozephalie. Sie weisen außerdem oft einen auffallend kurzen Nacken und einen kurzen Rumpf auf.
Der Morbus Hunter, der auch als Mukopolysaccharidose Typ II bezeichnet wird, ist eine X-chromosomal-rezessiv vererbte Erkrankung. Sie beruht auf einem Gendefekt auf dem X-Chromosom, das für das Enzym Iduronat-2-Sulfatase (I2S) kodiert, das den Abbau spezifischer Glykosaminoglykane (GAG) katalysiert. Durch die Mutation ist der GAG-Abbau gestört, das sich dadurch in den Zellen und Geweben anreichert. Es resultiert eine Multisystemerkrankung. Männer sind deutlich häufiger betroffen als Frauen.
Charakteristische Befunde sind häufige Infektionen der Atemwege und vor allem der Ohren, eine Nabel- oder Leistenhernie, Gelenkkontrakturen und ein vorgewölbter Bauch aufgrund einer Hepatomegalie. Es kommt außerdem oft zu Hautveränderungen mit Verdickungen und der Ausbildung weißlicher, knötchenartiger Läsionen. Zu beobachten ist meist eine Entwicklungsverzögerung der Kinder, insbesondere eine verzögerte Sprachentwicklung, Verhaltensauffälligkeiten wie beispielsweise eine Hyperaktivität und eine mentale Retardierung. Häufig tritt eine Gelenksteife auf, die zu unbeholfenen Bewegungen bis hin zu einem spastisch-ataktischen Gangbild führt. Auch eine Hör- und eventuell zusätzliche Visusstörung können beobachtet werden. Folgen können zudem eine Skoliose, eine Kyphose oder Kleinwuchs sein. Bleibt das Krankheitsbild unbehandelt, drohen erhebliche Komplikationen bis hin zu kardiovaskulären Folgeerkrankungen, die die Haupttodesursache dieser Patienten darstellen.
Allerdings ist auch der Morbus Hunter variabel in seiner Ausprägung. Es kann eine schwere Form mit geistiger Retardierung (früher Typ A) vorliegen, aber auch eine milde Krankheitsform mit geringer oder kaum merkbarer geistiger Entwicklungsverzögerung (früher Typ B). Dabei sind die Übergänge fließend.
Im Verdachtsfall ist die Bestimmung der GAG-Ausscheidung im Urin ratsam. Die Diagnose kann anschließend durch den Nachweis einer erniedrigten oder fehlenden Enzymaktivität im Serum gestellt werden. Ergänzend ist das defekte Enzym in Leukozyten oder Fibroblasten zu bestimmen. Möglich ist zudem eine molekulargenetische Analyse, ebenso wie eine pränatale Diagnose in Amnion- oder Chorionzottenzellen.
Zwar ist der Morbus Hunter nicht heilbar, aber die Erkrankung ist durch eine Enzymersatztherapie durchaus behandelbar. Dabei wird das Enzym Idursulfase einmal pro Woche als intravenöse Infusion verabreicht, was zu einer Stabilisierung des Krankheitsbildes führt. Dennoch ist die Prognose der Patienten limitiert, ihre Lebenserwartung ist stark eingeschränkt und die Mehrzahl der Betroffenen verstirbt bereits im zweiten Lebensjahrzehnt.
Christine Vetter
Merkenicher Str. 224, 50735 Köln

Christine Vetter
Merkenicher Straße 224,
50975 Köln


{kind=link}
{kind=link}