Das Pyoderma gangraenosum
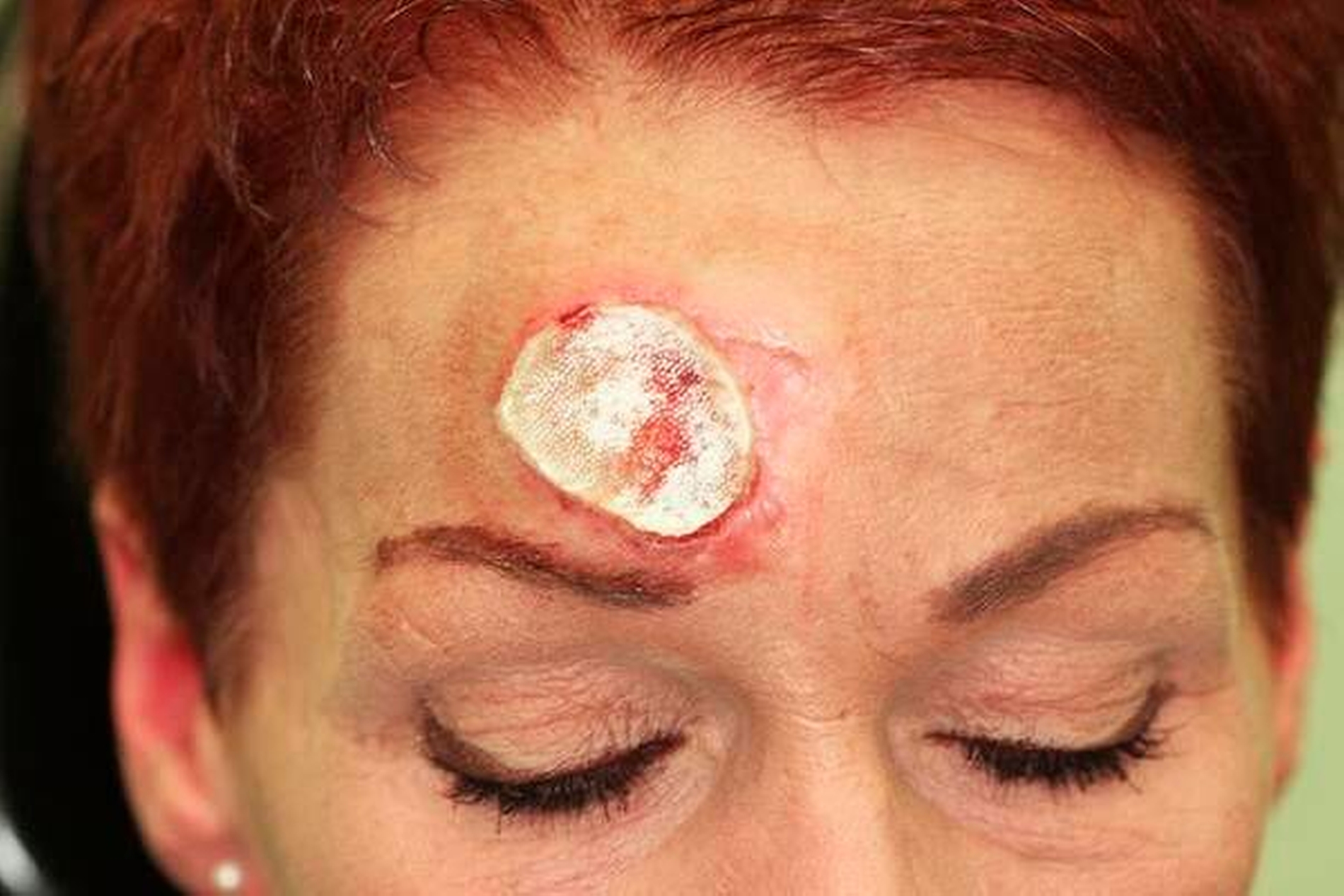
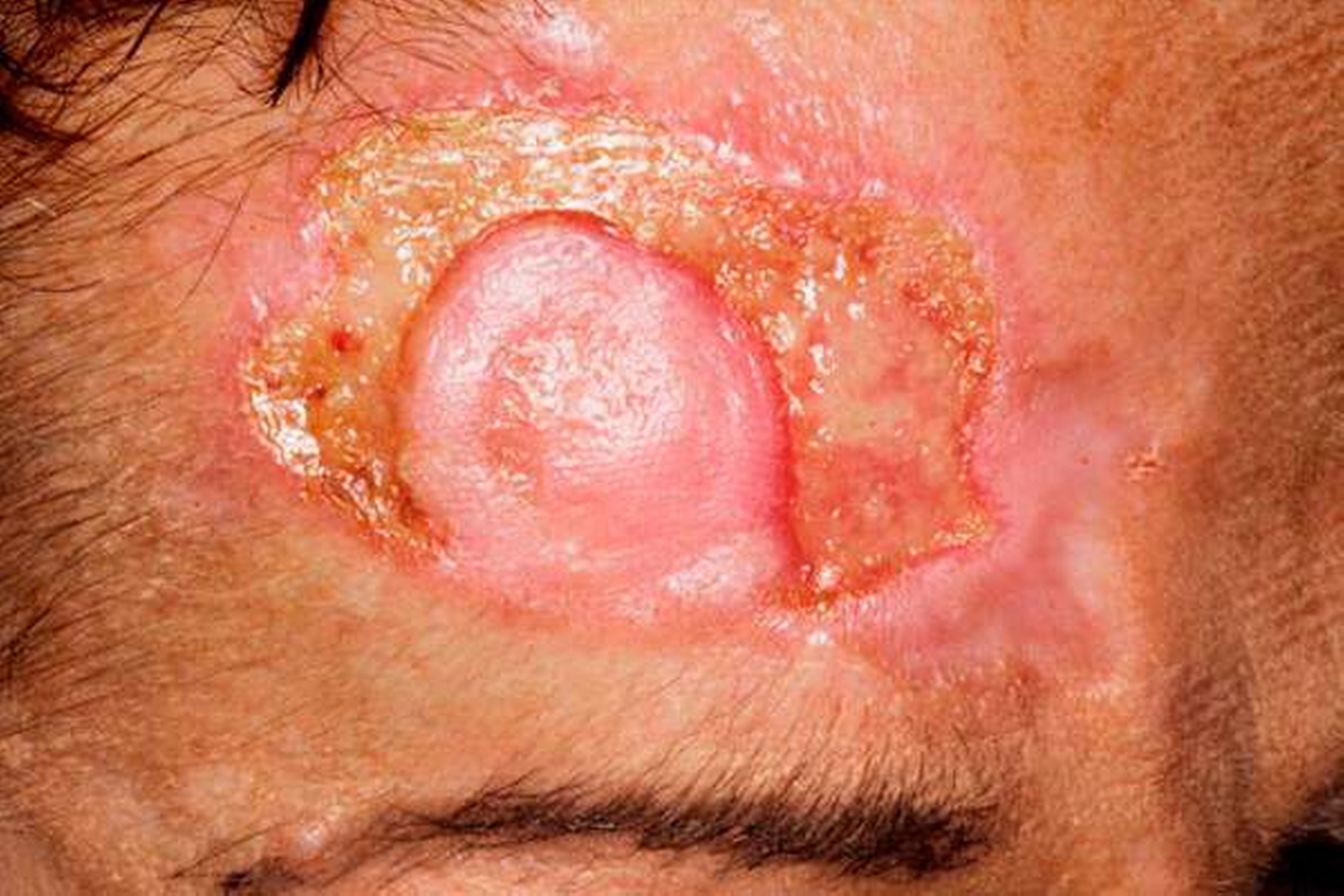
- Abbildung 1: Erstvorstellung im Oktober 2015: Die Läsion war teilweise mit Fibrin bedeckt, teils narbig ausgeheilt. © Fotos: Stegmann



Eine 47-jährige Patientin wurde in der Klinik und Poliklinik für Mund-, Kiefer- und Gesichtschirurgie der Uniklinik RWTH Aachen konsiliarisch zur Mitbeurteilung einer nicht abheilenden Wunde supraorbital rechts vorgestellt.
Anamnese und Befund
Inspektorisch war ein etwa 3 cm x 2 cm großer, kreisrunder Defekt mit aufgeworfenem, inflammatorisch verändertem Randsaum zu sehen. Die Läsion war teilweise mit Fibrin bedeckt und teils narbig ausgeheilt (Abbildung 1). Eigenanamnestisch bestand die Raumforderung seit etwa einem Jahr. Mehrfach seien bereits – frustrane – Wundrevisionen und Probebiopsien durchgeführt worden.
Durch alio loco durchgeführte Abstriche waren im Vorfeld Mykosen und atypische Mykobakteriosen ausgeschlossen worden. Auf der gesamten Gesichtshaut zeigten sich kleine lakunäre Öffnungen, teils narbig verheilt. Die Patientin gab weiter an, dass sie seit mehreren Jahren an rezidivierenden papulomatösen Läsionen, vor allem im Gesicht, leide.
Intraoral wurden im Wangen- und Lippenbereich zusätzlich zwei etwa 3 mm große aphthöse Läsionen festgestellt. Auf Nachfrage gab die Patientin an, dass diese schmerzhaften Veränderungen ab und zu auftreten würden.
Ansonsten wurde eine gute Mundhygiene ohne harte oder weiche Beläge befundet. Neben intermittierenden, rechts betonten Kopfschmerzen leide sie auch seit etwa vier Jahren an rezidivierenden Diarrhoen. Auch Myalgien der Extremitäten sowie ein Karpaltunnel-Syndrom beidseits wurden angegeben. Weiterhin liegen im Rahmen der Familienanamnese diverse Erkrankungen autoimmuner Genese (Multiple Sklerose und M. Hashimoto) vor.
Klinische und histologische Diagnosestellung
Das typische klinische Bild eines Pyoderma gangraenosums (PG) beginnt mit einem kleinen, geröteten Infiltrat, das sich rasch zu einer schmerzhaften Nekrose mit Ulzerationen ausdehnen kann und häufig einen bläulich-lividen, ödematös aufgeworfenen und gangränös unterminierten Rand aufweist. Sich ähnlich darstellende Erkrankungen wie Infektionen, maligne Entartungen oder Vaskulitiden müssen durch histopathologische und laborchemische Untersuchungen ausgeschlossen werden.
Ein PG ist klinisch, histologisch und immunhistologisch zu diagnostizieren. Bei unserer Patientin waren die immunhistologischen Untersuchungen (ANA, ANCA, RF) sowie eine Blutuntersuchung unauffällig. Zudem konnte durch eine Probeexzision mit ausreichend Gewebe neben der Ulzeration eine maligne Entartung ausgeschlossen werden.
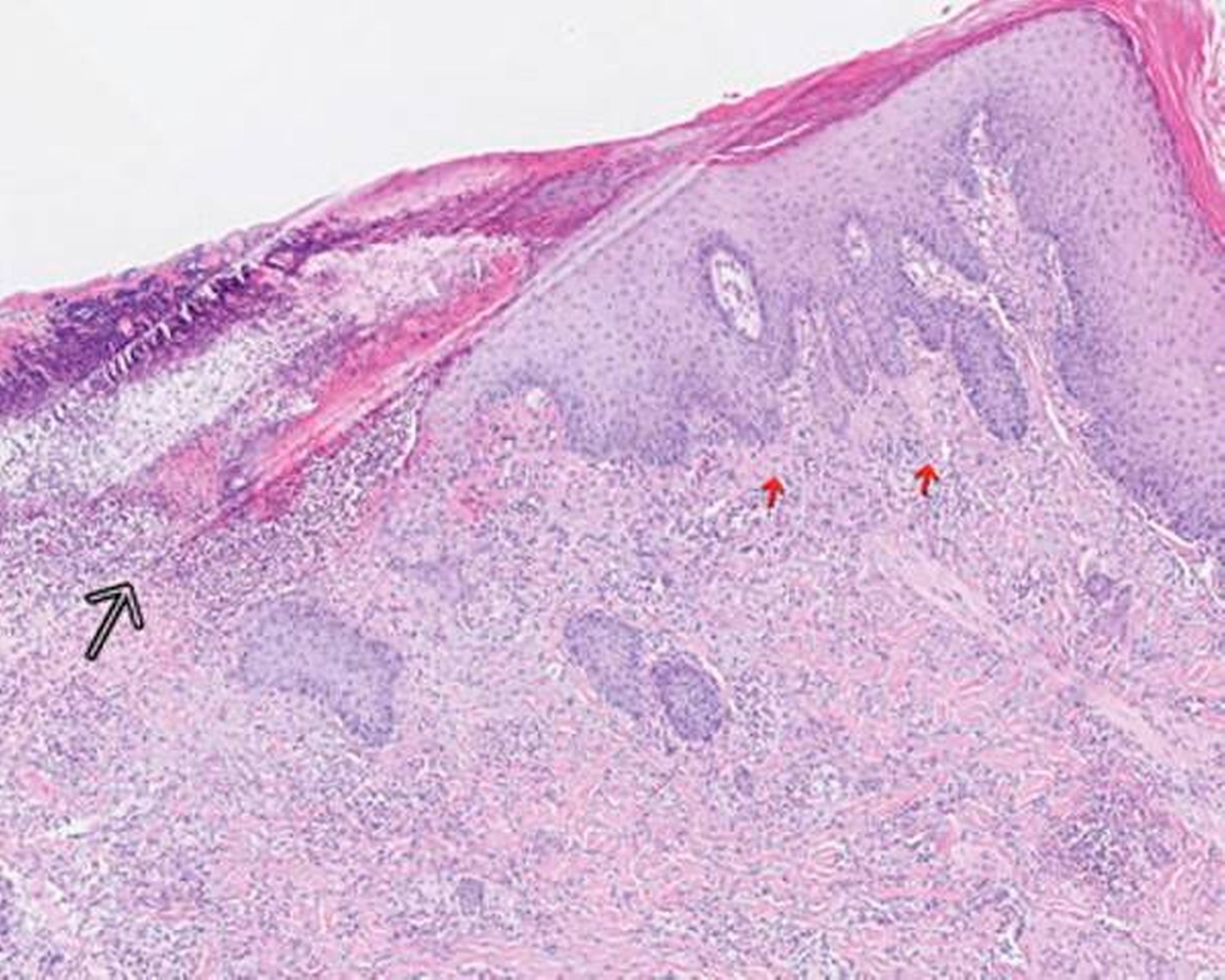
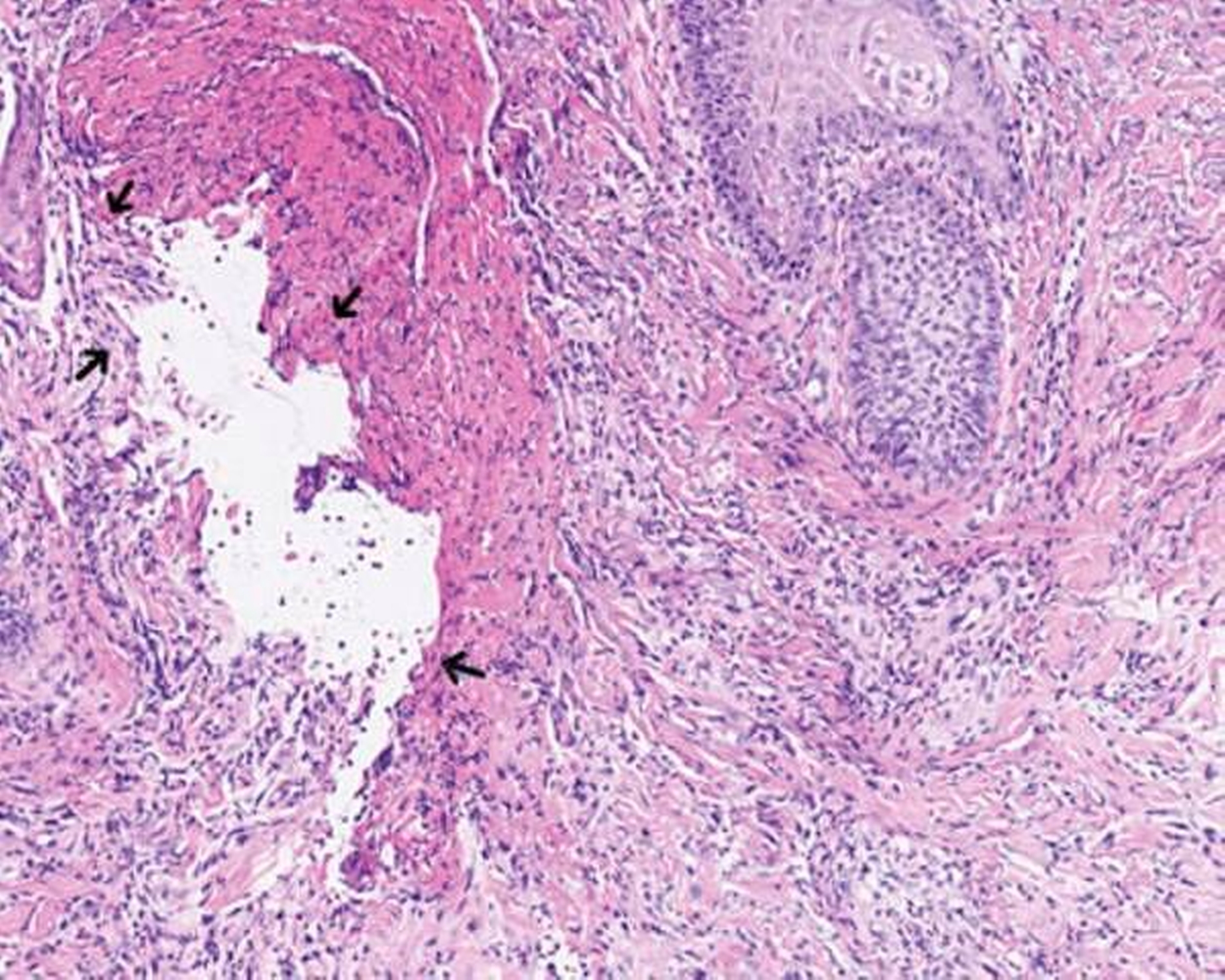
Mikroskopisch zeigten sich eine floride Ulzeration mit Fibrinexsudation und perivaskulären, entzündlichen Infiltraten mit neutrophilen Granulozyten sowie reaktive Veränderungen der Epidermis (Abbildungen 2 und 3). Auch bei einem erneuten Abstrich konnten keine Bakterien nachgewiesen werden.
Therapie und Verlauf
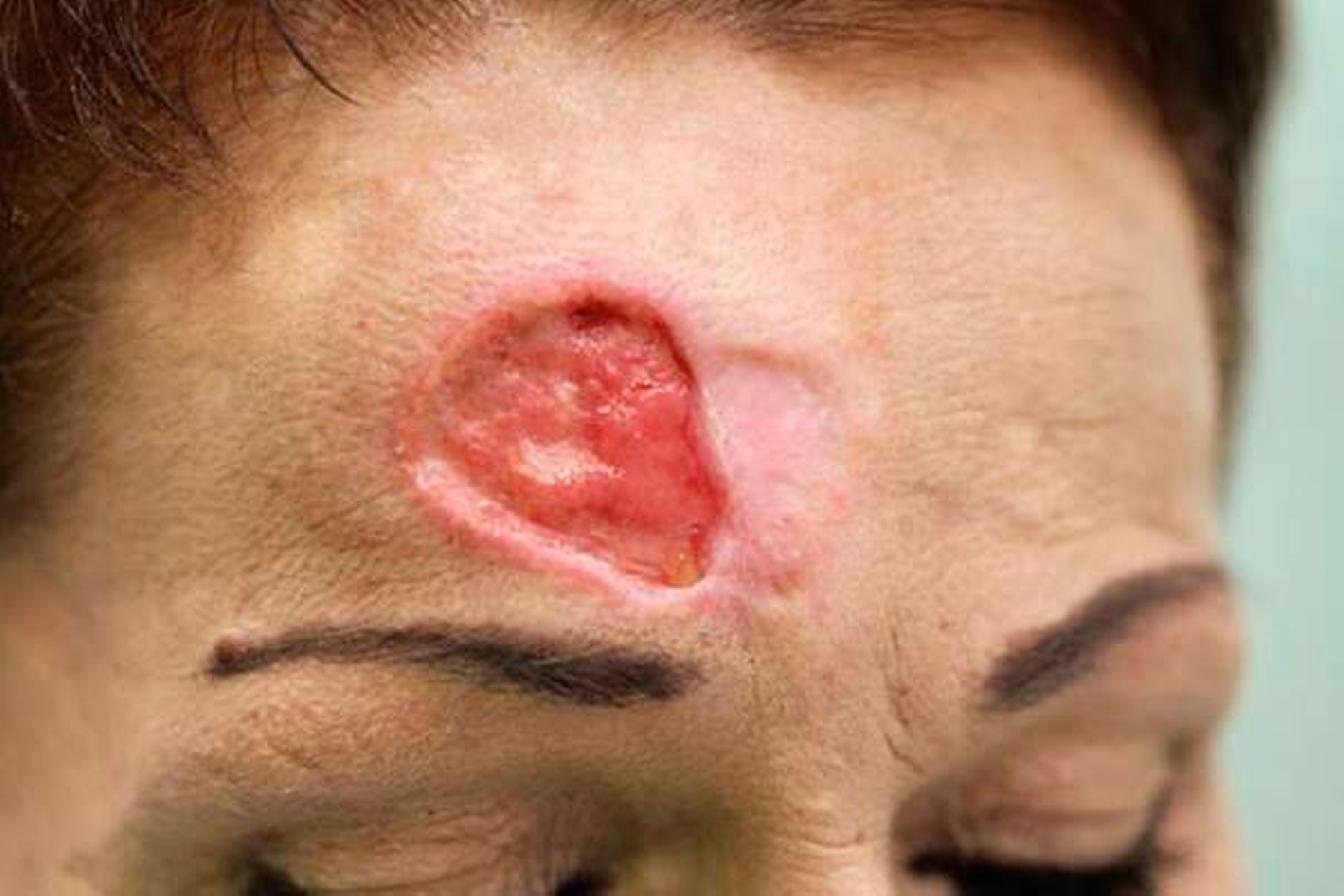
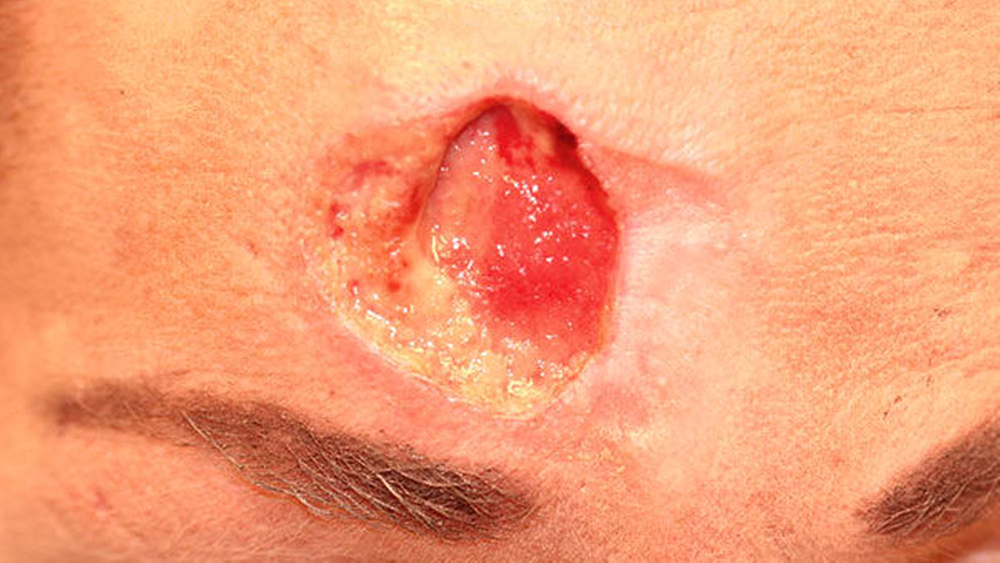
Nach lokaler chirurgischer Wundanfrischung mit Entnahme einer Probebiospie und regelmäßigen Verbandswechseln zeigte sich im Verlauf keine Abheilungstendenz der Läsion. Vielmehr nahm diese an Größe noch zu. (Abbildungen 4 und 5).
Nach weiteren chirurgischen Interventionen in Form von Wundanfrischungen und erneuten Probeexzisionen wurde die Verdachtsdiagnose eines Pyoderma gangraenosum gestellt. Andere relevante Differenzialdiagnosen wurden durch die bereits gewonnenen histologischen und bakteriologischen Untersuchungen ausgeschlossen, so dass umgehend eine Hochdosis-Kortison- Therapie (Prednisolon 80 mg bis zur Abheilung) eingeleitet wurde.
Unter dieser systemischen Therapie und lokaler Wundpflege mit nicht haftenden Fettgazeverbänden konnte eine zunehmende Abheilung der Läsion beobachtet werden (Abbildungen 6 und 7).
Diskussion
Das PG ist eine seltene neutrophile Dermatose, die sich durch sterile Hautulzera mit einem unterminierenden erythematösen Randsaum manifestiert [Alavi et al., 2017; Cugno et al., 2017; Androutsakos et al., 2015; Su et al., 2004; Powell et al., 1996]. Die Erkrankung betrifft beide Geschlechter und jedes Alter.
Die unkontrollierte kutane Ausbreitung verläuft individuell unterschiedlich. Die Pathophysiologie ist sehr komplex und noch nicht vollständig verstanden. Aktuell geht man von einer autoimmunen Genese aus [Alavi et al., 2017]. Allgemein wird angenommen, dass eine verstärkte immunologische Reaktion und eine Überproduktion von Interleukinen (IL-1 Interleukine) stattfinden [Cugno et al., 2017].
Die nicht-infektiöse Pathogenese geht mit einer Fehlfunktion der neutrophilen Granulozyten einher [Su et al., 2004; Wollina et al., 2007]. Klinisch zeigen sich beim PG schmerzhafte, nekrotische Hautulzera mit einem irregulär unterminierenden Randsaum und rascher Progression [Su et al., 2004].
Die Diagnose stützt sich maßgeblich auf das klinische Bild mit einer sterilen Ulzeration und typischem Aussehen. Ein einfacher Test zur Bestätigung der Diagnose liegt nicht vor. Differenzialdiagnosen (Erysipel, nekrotisierende Vaskulitis, tumoröse Manifestationen, Hauttuberkulose, Lues und mehr) müssen durch spezifische Tests ausgeschlossen werden.
Eine Probeexzision im Frühstadium ist hilfreich, da sie den Ausschluss einer Vaskulitis der Haut erlaubt und gegebenenfalls eine lymphozytäre Infiltration nachgewiesen werden kann. Außerdem kann so eine maligne Läsion ausgeschlossen werden.
Im Spätstadium zeigt sich histologisch eine Infiltration mit neutrophilen Granulozyten sowie Hämorrhagien [Su et al., 1986; Callen et al., 1998].
In der Serologie lassen sich keine spezifischen Veränderungen nachweisen. Beschrieben sind jedoch unter anderem monoklonale Gammopathien und der Nachweis variabler Autoantikörper. Das PG ist in 50 bis 70 Prozent der Fälle mit systemischen Erkrankungen einschließlich chronisch-entzündlichen Darmerkrankungen, rheumatoider Arthritis und lymphoproliferativen Erkrankungen assoziiert. Deren Abklärung sollte fester Bestandteil der Diagnostik sein [Androutsakos et al., 2015; Wayte et al., 1995; Török et al., 2000; Powel et al.,1985; Stolman et al.,1975; Bernstein et al., 2001].
Auch die Therapie muss für jeden Patienten je nach Lokalisation, Anzahl und Größe der Läsionen, der extrakutanen Manifestationen und weiterer assoziierter Erkrankungen wie PAPA (pyogenic arthritis, PG and acne), PASH (PG, acne and suppurative hidradenitis) oder PAPASH (pyogenic arthritis, acne, PG and suppurative hidradenitis) spezifisch angepasst werden [Cugno et al., 2017]. Nebenwirkungen der immunsuppressiven Therapie sowie Komorbiditäten müssen berücksichtigt werden. Besonders bei akuten und schnell wachsenden Prozessen wird eine zeitnahe systemische Therapie empfohlen [Alavi et al., 2017].
Die lokale Therapie ist nicht-chirurgisch und besteht aus der Wundpflege vorzugsweise mit nicht-haftenden Fettgazen. Kürettagen können in einigen Fällen die Wundheilung fördern. Operative Maßnahmen (wie mit Wunddebridement und Nekrosenabtragung) sind kontraindiziert, da sie als Trigger zu einer Verschlechterung und Größenzunahme der Läsion führen können.
Neben den topischen und intraläsionalen Therapieoptionen, die im Frühstadium und bei milden Verlaufsformen teilweise ausreichend sein können, sind bei fortgeschrittenen Defekten vorzugsweise systemische Therapieansätze erfolgsversprechend. Die Studienlage zeigt für systemische Gaben von Kortison-haltigen Präparaten und Cyclosporin A sowie Biologicals wie den TNF-α-Inhibitor Infliximab aktuell die besten Ergebnisse.
Zusätzliche Therapieoptionen stellen die TNF-α-Inhibitoren Adalimumab und Etanercept, der Interleukin(IL)-12/23-Antikörper Ustekinumab, der IL-1-Rezeptor-Antagonist Anakinra und der IL-1α-Antikörper Canakinumab dar [Quist et al., 2017].

Julius Steegmann
Dr. Alexander Bartella
Prof. Dr. Dr. Frank Hölzle
PD Dr. Dr. Timm Steiner
Klinik für MKG-Chirurgie
Uniklinik RWTH Aachen,
Pauwelsstr. 30 , 52074 Aachen,
jsteegmann@ukaachen.de
Prof. Jens-Malte Baron
Klinik für Dermatologie und Allergologie
Hautklinik der Uniklinik RWTH Aachen,
Pauwelsstr. 30, 52074 Aachen
Dr. med. Claudio Cacchi
Institut für Pathologie,
Uniklinik RWTH Aachen,
Pauwelsstr. 30, 52074 Aachen
Fazit für die Praxis
Bei nicht abheilenden intra- oder extraoralen Wunden müssen systemische Erkrankungen interdisziplinär abgeklärt werden
Chirurgische Interventionen sind bei Diagnosestellung eines Pyoderma gangraenosums kontraindiziert.
Bei anamnestischen Auffälligkeiten im Kopf-Hals-Bereich ist eine sorgfältige orale Untersuchung nicht erst zum Ausschluss anderer Ursachen, sondern bereits im Rahmen der Primärdiagnostik empfehlenswert.
Literaturliste
(Am J Clin Dermatol. 2017 Feb 21. doi: 10.1007/s40257–017–0251–7. [Epub ahead of print] Pyoderma Gangrenosum: An Update on Pathophysiology, Diagnosis and Treatment. Alavi A1, French LE2, Davis MD3, Brassard A4, Kirsner RS5.)
BMC Gastroenterol. 2015 Oct 26;15:149. doi: 10.1186/s12876–015–0376–1. A case report of successful treatment of pyoderma gangrenosum in a patient with autoimmune hepatitis, and review of the literature. Androutsakos T1, Stamopoulos P2, Aroni K3, Hatzis G4.
Bernstein CN, Blanchard JF, Rawsthorne P, et al. The prevalence of extraintestinal diseases in inflammatory bowel disease: a population based study. Am J Gastroenterol. 2001;96:1116–22. doi: 10.1111/j.1572–0241.2001.03756.x.
Callen JP. Pyodermagangrenosum. Lancet. 1998;351:581–5. doi: 10.1016/S0140–6736(97)10187–8.
(Am J Clin Dermatol. 2017 Feb 25. doi: 10.1007/s40257–017–0265–1. [Epub ahead of print] PAPA, PASH and PAPASH Syndromes: Pathophysiology, Presentation and Treatment. Cugno M1, Borghi A2, Marzano AV3.
Powell FC, Schroeter AL, Su WP, et al. Pyoderma gangrenosum: a review of 86 patients. Q J Med. 1985;55:173–86.
Powell FC, Su WP, Perry HO. Pyodermagangrenosum classification and management. J Am Acad Dermatol. 1996;34:395–409. doi: 10.1016/S0190–9622(96)90428–4.
J Dtsch Dermatol Ges. 2017 Jan;15(1):34–40. doi: 10.1111/ddg.13173. Treatment options for pyoderma gangrenosum. Quist SR1, Kraas L1.
Stolman LP, Rosenthal D, Yaworsky R, et al. Pyoderma gangrenosum and rheumatoid arthritis. Arch Dermatol. 1975;111:1020–3. doi: 10.1001/archderm.1975.01630200080009.
Su WP, Schroeter AL, Perry HO, Powell FC. Histopathologic and immunopathologic study of pyodermagangrenosum. J CutanPathol. 1986;13:323–30.
Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria [review] Int J Dermatol. 2004;43:790–800. doi: 10.1111/j.1365– 4632.2004.02128
Török L, Kirschner A, Gurzo M, et al. Bullous pyoderma gangrenosum as a manifestation of leukemia cutis. Eur J Dermatol. 2000;10:463–5.
Wayte JA, Rogers S, Powell FC. Pyoderma gangrenosum, erythema elevatum diutinum and IgA monoclonal gammopathy. Australas J Dermatol. 1995;36:21–3. doi: 10.1111/j.1440– 0960.1995.tb00919.x.
Wollina U. Pyodermagangrenosum—a review. Orphanet J Rare Dis. 2007;2:19. doi: 10.1186/1750– 1172–2–19
Julius Steegmann
Pauwelsstr. 30,
52074 Aachen

Univ.-Prof. Dr. med. Dr. med. dent. Frank Hölzle
Klinik und Poliklinik für Mund-,
Kiefer- und Gesichtschirurgie,
Universitätsklinikum RWTH Aachen
Pauwelsstr. 30, 52074 Aachen


{kind=link}
{kind=link}