Diagnose einer familiären adenomatösen Polyposis coli
- © LMU München

Ein elfjähriges Mädchen stellte sich nach Überweisung ihres Hauszahnarztes aufgrund einer seit drei Monaten langsam größenprogredienten Raumforderung im Bereich des basalen Kieferwinkels links in unserer Poliklinik vor. Aktuell befand sich die Patientin in kieferorthopädischer Behandlung aufgrund multipler Zahnfehlstellungen und Retentionen. Seit der Geburt seien zudem multiple histologisch gesicherte epidermale Zysten im Bereich der behaarten Kopfhaut und der Extremitäten aufgetreten.
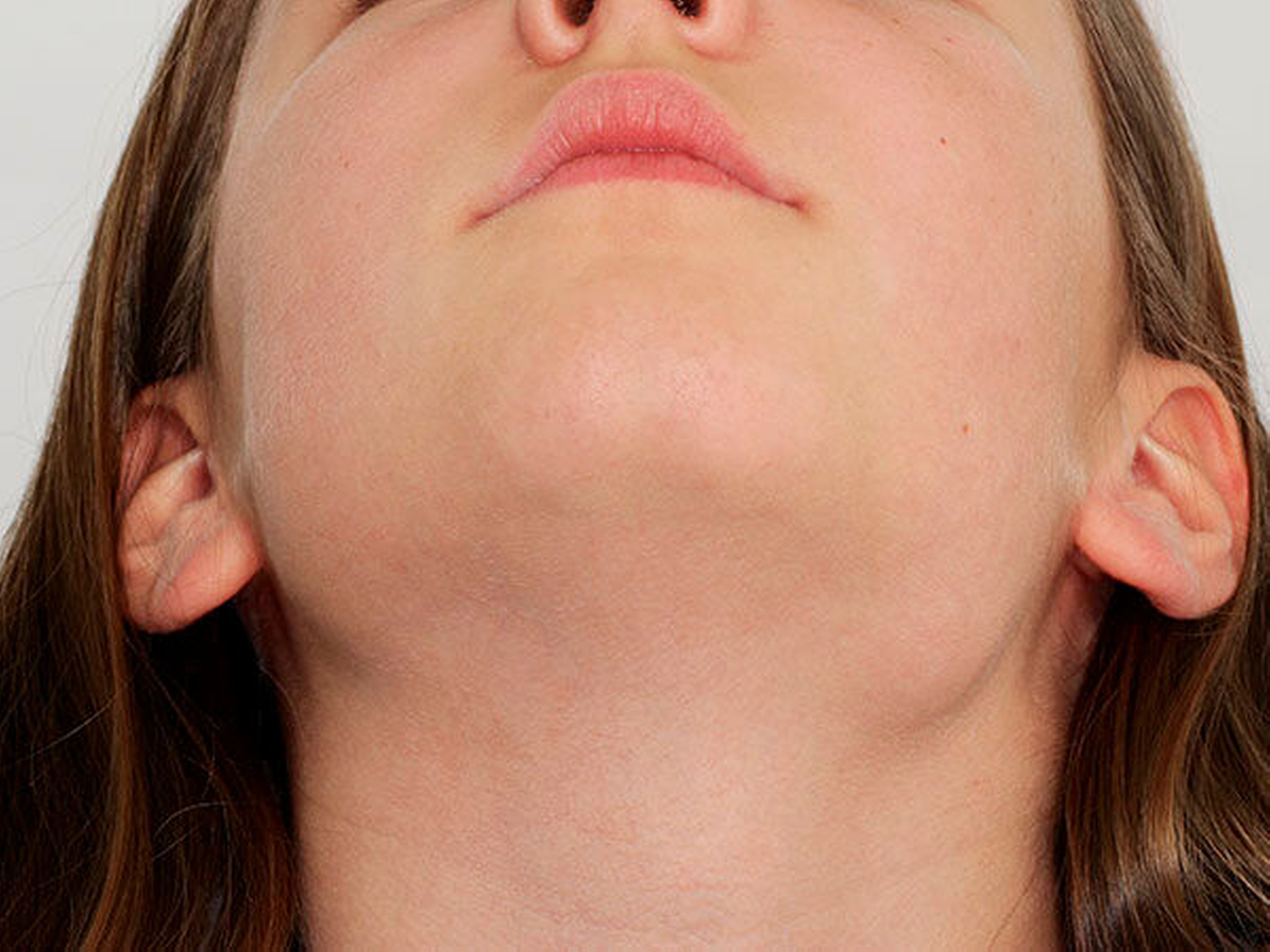
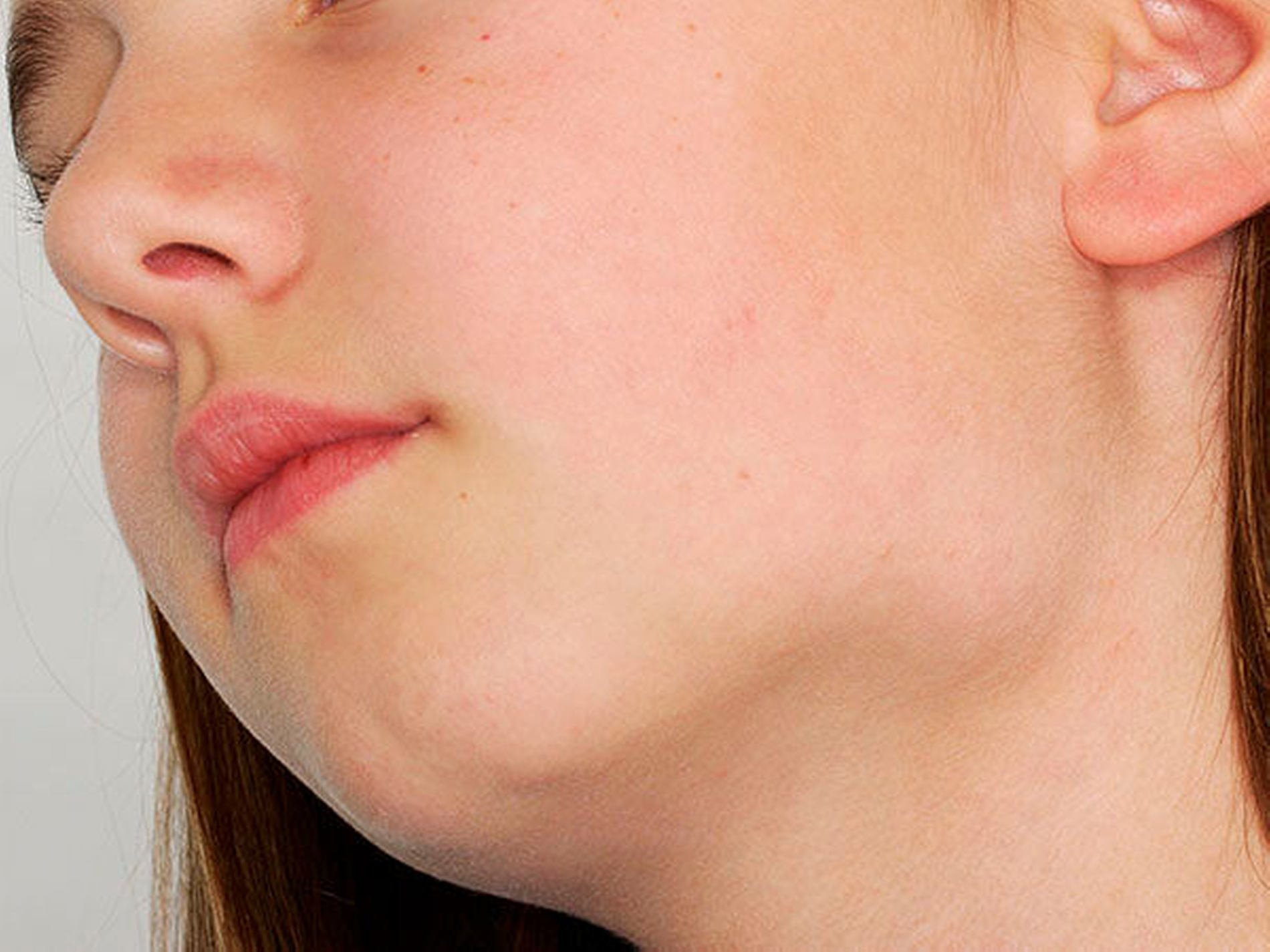
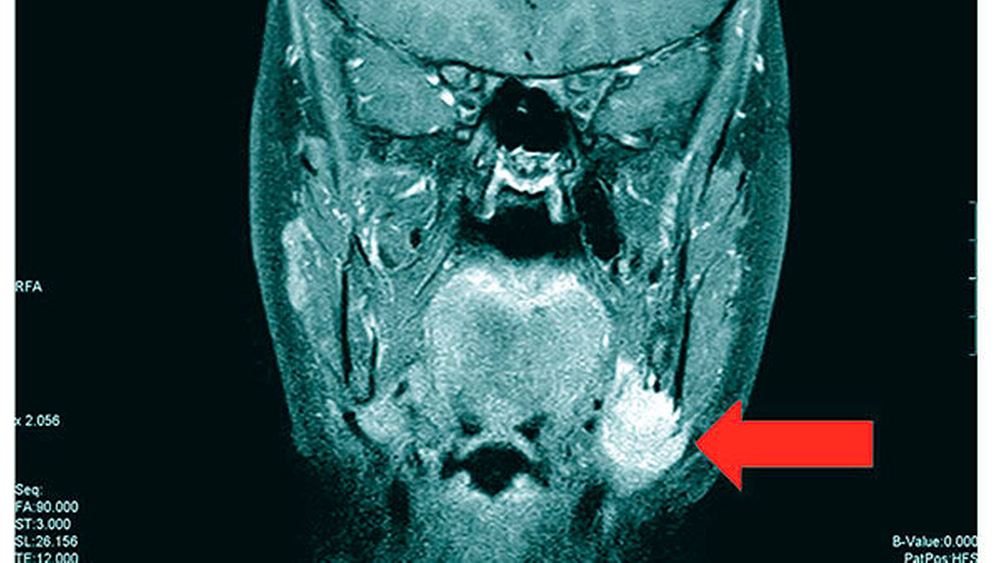
Bei der klinischen Untersuchung zeigte sich im Bereich des basalen Kieferwinkels links eine knöcherne, 3 cm x 2 cm messende Raumforderung, die nicht druckschmerzhaft oder verschieblich war. (Abbildung 1). Palpatorisch zeigten sich im Bereich der behaarten Kopfhaut multiple subkutane Raumforderungen, die weich, nicht druckschmerzhaft sowie verschieblich waren und eine maximale Ausdehnung von 3 cm x 3 cm aufwiesen.
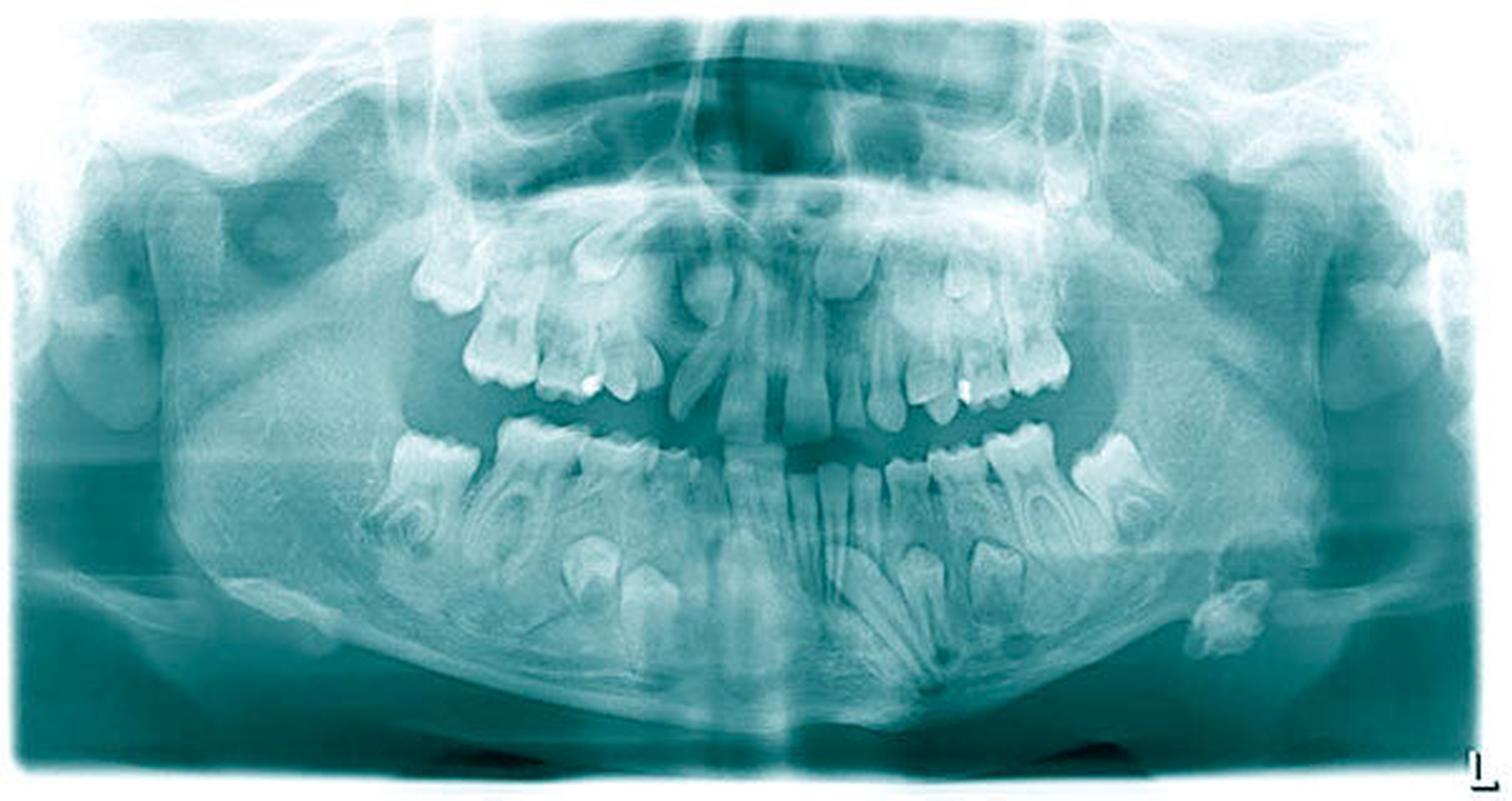
Die durchgeführte Panoramaschichtaufnahme zeigte eine knöcherne Apposition im Bereich des Kieferwinkels links basal mit einer Ausdehnung von 2 cm x 2 cm. Es bestand eine Retention der Zähne 15, 13, 23, 25, 27, 35, 34, 33, 43, 44 und 45 sowie die Nichtanlage des Zahns 22. Die Milchzähne 55, 52, 63, 65, 73 und 83 persistierten. Radiologisch zeigten Maxilla und Mandibula milchglasartige Strukturauffälligkeiten (Abbildung 2).
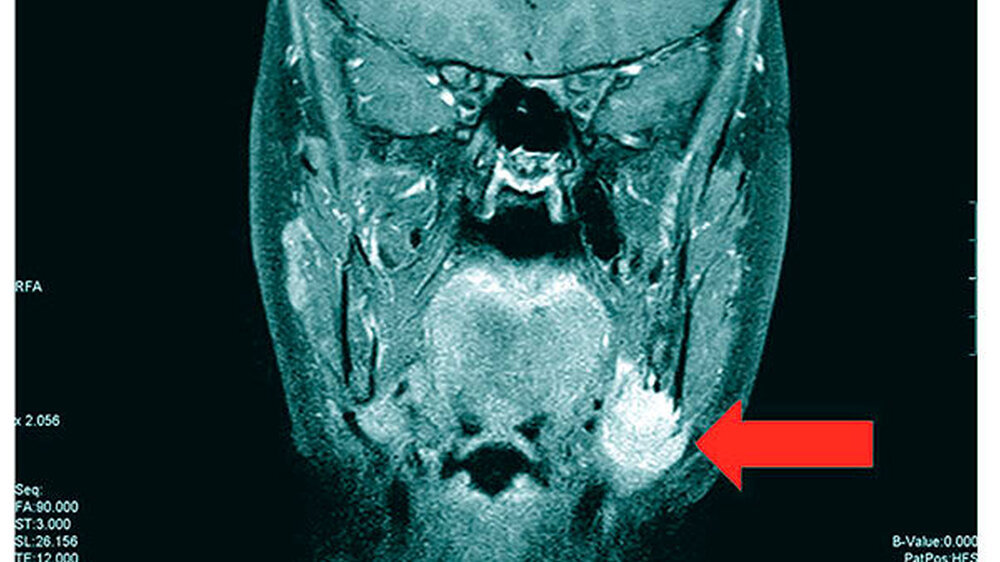
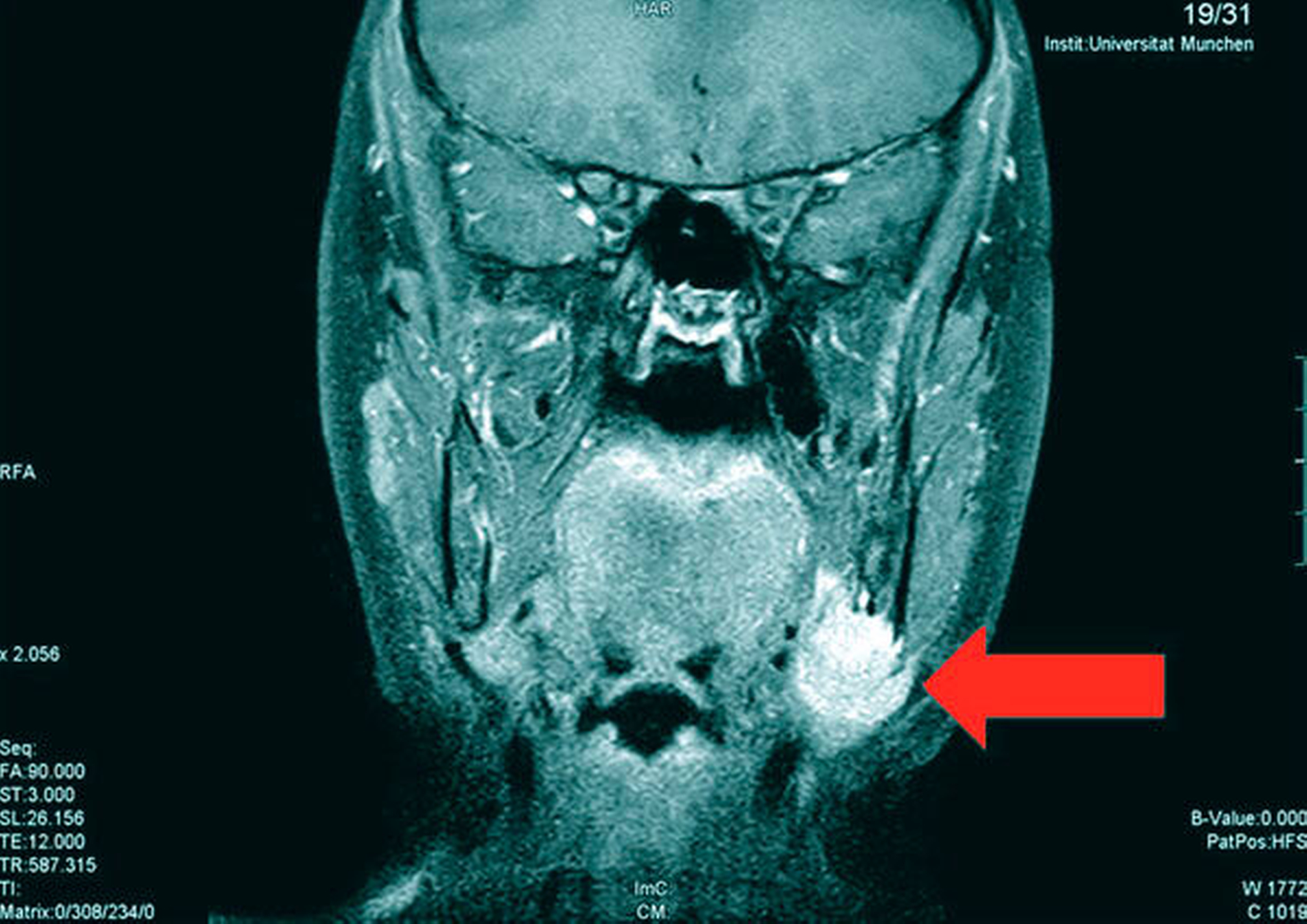
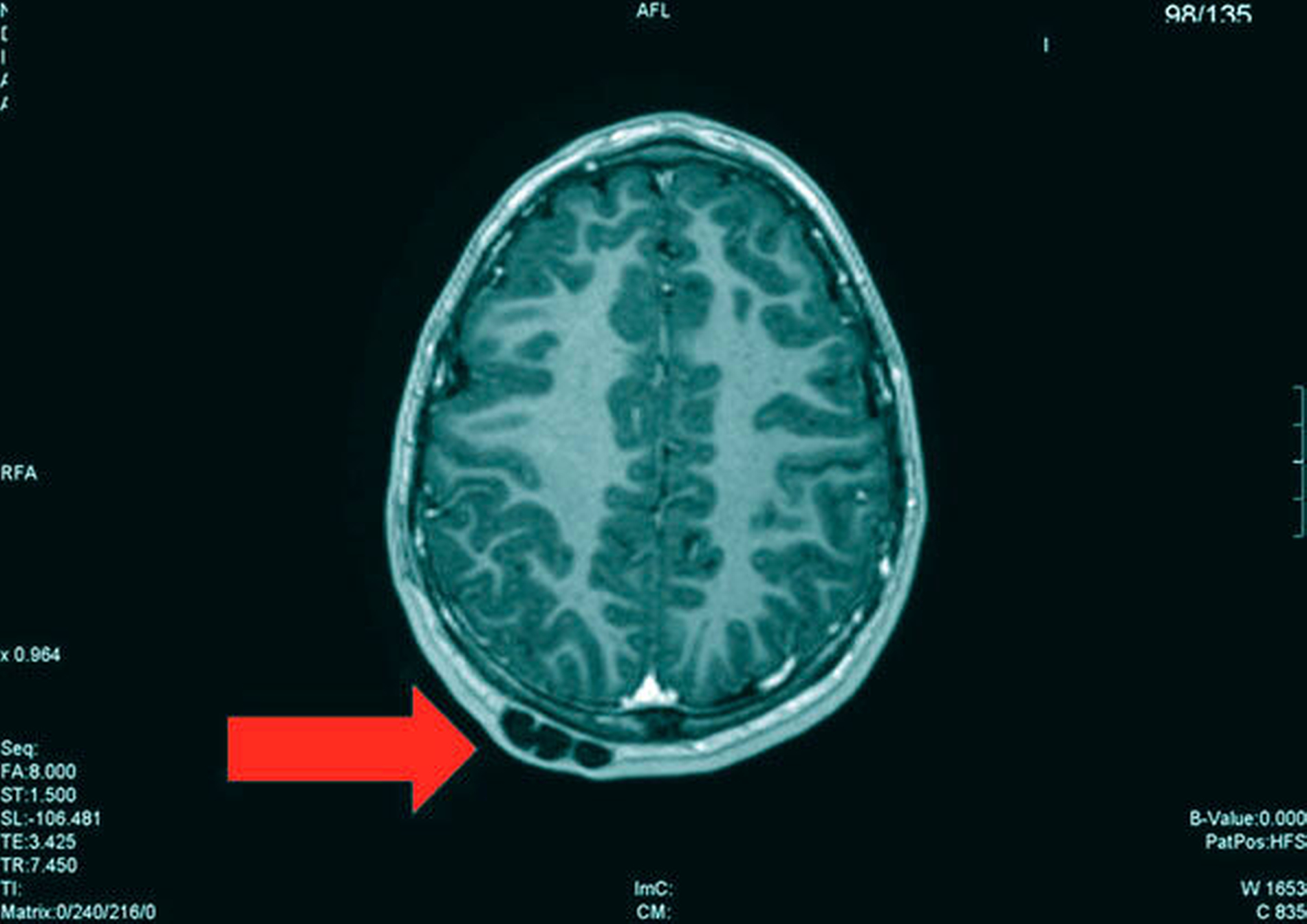
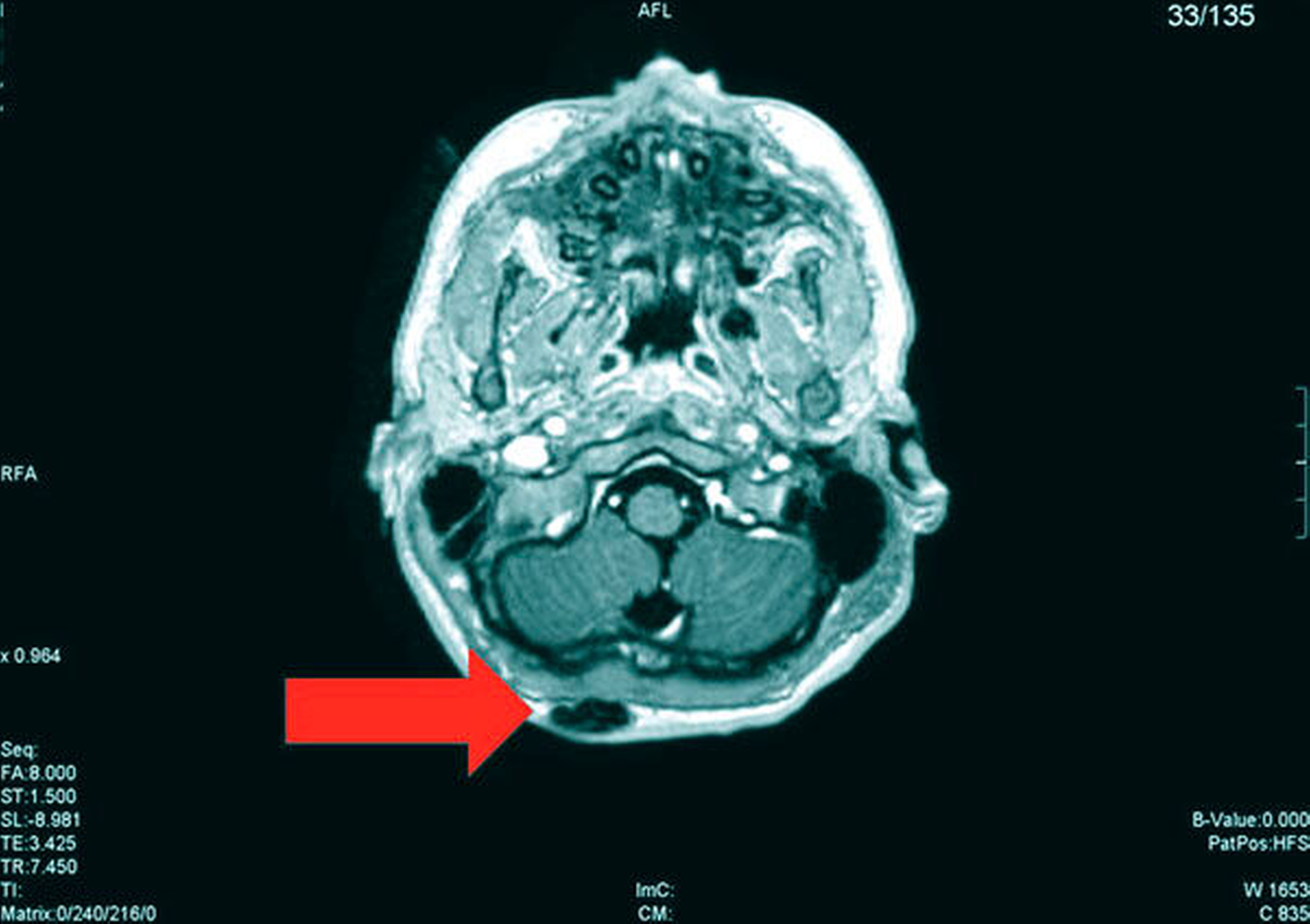
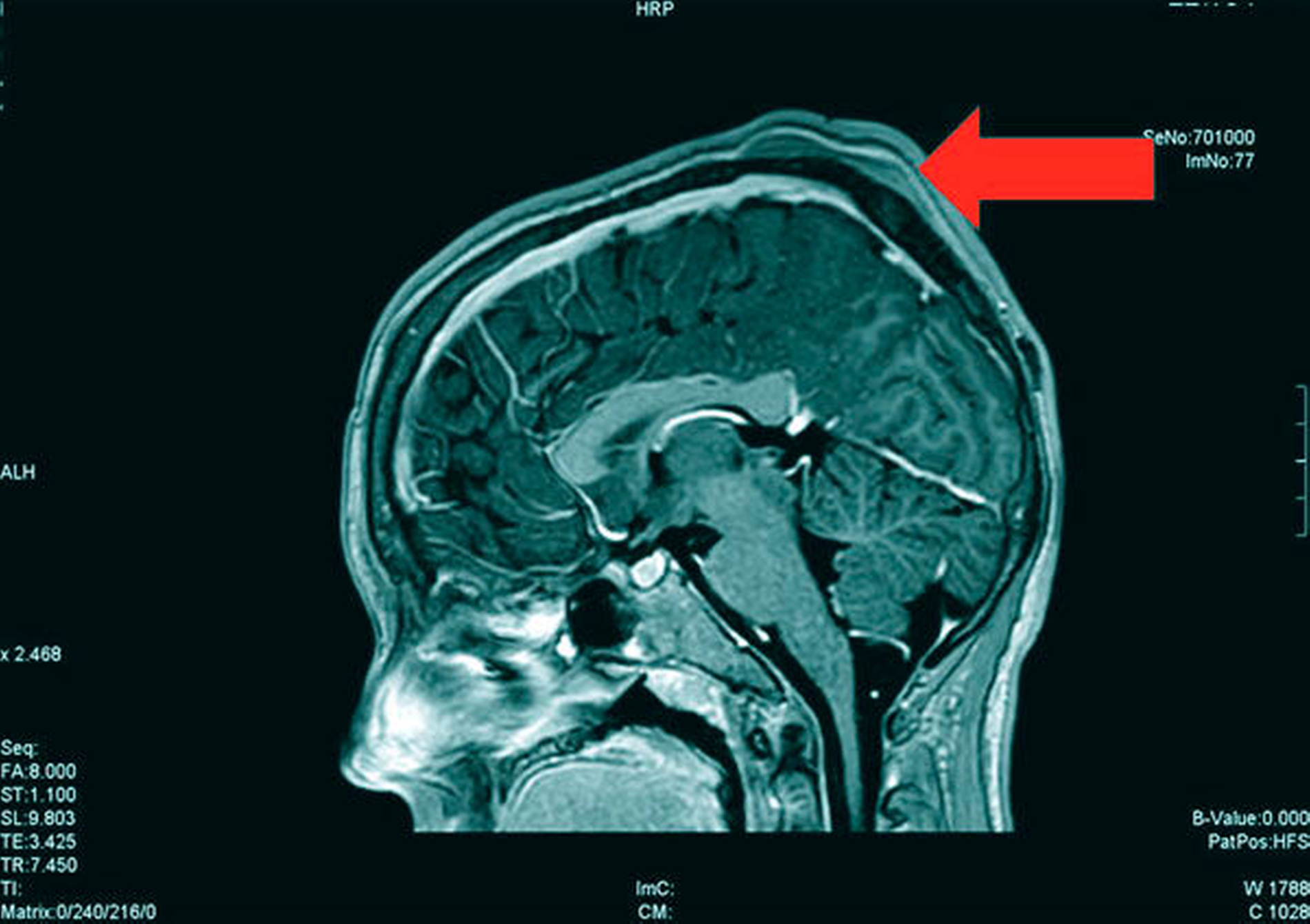
Die Magnetresonanztomografie des Neuro- und des Viszerokraniums erbrachte den Nachweis eines Areals erhöhter Kontrastmittelaufnahme im Bereich des basalen Kieferwinkelrandes links mit einer Ausdehnung von 3,1 cm x 2,3 cm. Im Bereich der behaarten Kopfhaut zeigten sich subkutan T1-hypointense, zum Teil polylobulierte Läsionen mit Verdacht auf epidermale Zysten sowie subgaleale T2-hypointense, flächig kontrastmittelaufnehmende Läsionen mit Verdacht auf Osteome (Abbildungen 3a bis 3d).
Therapie
In Intubationsnarkose erfolgte zunächst die Exzisionsbiopsie im Bereich des basalen Kieferwinkels links über einen transoralen Zugang. Eine operative Entfernung der Raumforderungen im Bereich der Kopfhaut wurde abgelehnt. Der postoperative Verlauf gestaltete sich nach Rückgang der initial ausgeprägten Schwellung komplikationslos.
Die Analyse des intraoperativ gewonnenen Materials zeigte vitales Knochengewebe bestehend aus teils Lamellen- und Geflechtknochen mit verstärkten An- und Umbauvorgängen. Ein Anhalt für Spezifität oder Malignität bestand nicht. Klinisch erfolgte die Diagnose eines Osteoms.
Aufgrund des histologisch gesicherten Osteoms sowie des anamnestisch vorbekannten Auftretens multipler epidermaler Zysten, der Retention multipler Zähne und der milchglasartigen Knochenstrukturveränderung des Ober- und Unterkiefers erfolgte die Vorstellung der Patientin in der humangenetischen Sprechstunde. Bei Verdacht auf das Vorliegen einer familiären adenomatösen Polyposis coli (FAP) im Rahmen eines Gardner-Syndroms wurde eine APC-Gendiagnostik durchgeführt. Diese zeigte eine krankheitsrelevante heterozygote Mutation.
Veranlasst wurde die Durchführung einer diagnostischen Gastro-Duodenoskopie sowie einer Koloskopie. Hierbei zeigten sich im Bereich der Magenschleimhaut multiple sessile Polypen ohne histologische Malignitätskritierien. Der Befund der Koloskopie war unauffällig. Zukünftig sind regelmäßige Darmtumor-Vorsorgeuntersuchungen und gegebenenfalls die Abtragung von Polypen lebenslang erforderlich. Wegen der Milchzahnpersistenzen, der Zahnretentionen und der radiologisch auffälligen Kieferknochenbefunde erfolgten in einem Zweiteingriff die Entfernung der persistierenden Milchzähne, die Insertion von palatinalen Minipins zur kieferorthopädischen Zahneinstellung und eine Knochenbiopsie. Letztere erbrachte den histologischen Nachweis zemento-ossärer Dysplasien.
Diskussion
Die FAP ist eine genetisch bedingte Tumorneigung mit einer Prävalenz von 1:11.300 bis 1:37.600 in der EU [Half et al., 2009]. Ursächlich für die FAP sind Veränderungen im APC-Gen, die autosomal dominant vererbt werden oder durch spontane Mutation entstehen können. Die FAP zeigt sich in erster Linie durch eine massive Ausbildung von Dickdarmpolypen. Die Polypen entstehen ab der Kindheit und sind bei Anlageträgern spätestens bis zum 40. Lebensjahr manifest. Dabei gibt es Hunderte bis Tausende Dickdarmpolypen, die bösartig entarten können. Bei FAP-Patienten tritt aufgrund der Vielzahl der Polypen unbehandelt praktisch immer ein Kolonkarzinom auf. Deshalb wird bei klassischer FAP die prophylaktische Entfernung des gesamten Dickdarms empfohlen. Ansonsten sind engmaschige, regelmäßige Vorsorgekontrollen durch Kolo- sowie Gastro-Duodenoskopien erforderlich [Half et al., 2009].
Bei einigen FAP-Patienten treten neben den Darmtumoren auch weitere, in der Regel überwiegend gutartige Tumore auf, beispielsweise Fibrome, Epidermoidzysten, Osteome und Lipome. In diesem Fall wird die Erkrankung als Gardner-Syndrom bezeichnet [Pereira et al., 2016]. Als charakteristische sonstige Symptome finden sich bei FAP-Anlageträgern auch Zahnanomalien oder Zahnanlagestörungen. Daneben weisen FAP-Anlageträger ein leicht erhöhtes Risiko für das Auftreten papillärer Schilddrüsenkarzinome, Hepatoblastome und Gallengangskarzinome auf. Weiterhin kommt es häufig zu einer typischen Augenhintergrundveränderung (kongenitale Hyperplasie des retinalen Pigmentepithels; CHRPE), die bereits im Kindesalter auf eine familiäre FAP-Anlageträgerschaft hinweisen kann [Galiatsatos et al., 2006; Half et al., 2009].
Fazit für die Praxis
Sollte es bei Patienten schon in kindlichem Alter zum Auftreten von multiplen benignen Tumoren und Zysten (Osteome, zemento-ossäre Dysplasien, Epidermoidzysten) in Kombination mit einer Retention und Nichtanlage von Zähnen kommen, ist eine humangenetische Vorstellung sinnvoll.
Dr. med. Dr. med. dent. Jakob Ihbe
Arzt in Weiterbildung und Zahnarzt
Klinik für Mund-, Kiefer- und Gesichtschirurgie
Klinikum der Universität München, Campus Innenstadt
Lindwurmstr. 2a, 80337 München
jakob.ihbe@med.uni-muenchen.de
PD Dr. med. Yasmin Mehraein
Institut für Humangenetik der LMU-München
Goethestr. 2980336 München
Yasmin.Mehraein@med.uni-muenchen.de
Prof. Dr. Dr. Michael Ehrenfeld
Direktor der Klinik für Mund-, Kiefer- und Gesichtschirurgie
Klinikum der Universität München, Campus Innenstadt
Lindwurmstr. 2a, 80337 München
michael.ehrenfeld@med.uni-muenchen.de
Literaturliste
Galiatsatos, P. and W.D. Foulkes (2006) Familial adenomatous polyposis. Am J Gastroenterol, 101(2): p. 385–98
Half, E., D. Bercovich and P. Rozen (2009) Familial adenomatous polyposis. Orphanet J Rare Dis, 4: p. 22.
Pereira, D.L., P.A. Carvalho, M.I. Achatz, A. Rocha, G. Tardin Torrezan and F.A. Alves (2016) Oral and maxillofacial considerations in Gardner‘s syndrome: a report of two cases. Ecancermedicalscience, 10: p. 623.

Dr. med. Jakob Ihbe
Klinik für Mund-, Kiefer- und Gesichtschirurgie
Klinikum der Universität München
Campus Innenstadt
Lindwurmstr. 2a, 80337 München
PD Dr. med. Yasmin Mehraein
Goethestr. 29
80336 München

Dr. Michael Ehrenfeld
Klinikum der Universität München
Campus Innenstadt
Lindwurmstr. 2a
80337 München


{kind=link}
{kind=link}