Orbitaler und periorbitaler Tumor: Das sphenoorbitale Meningeom
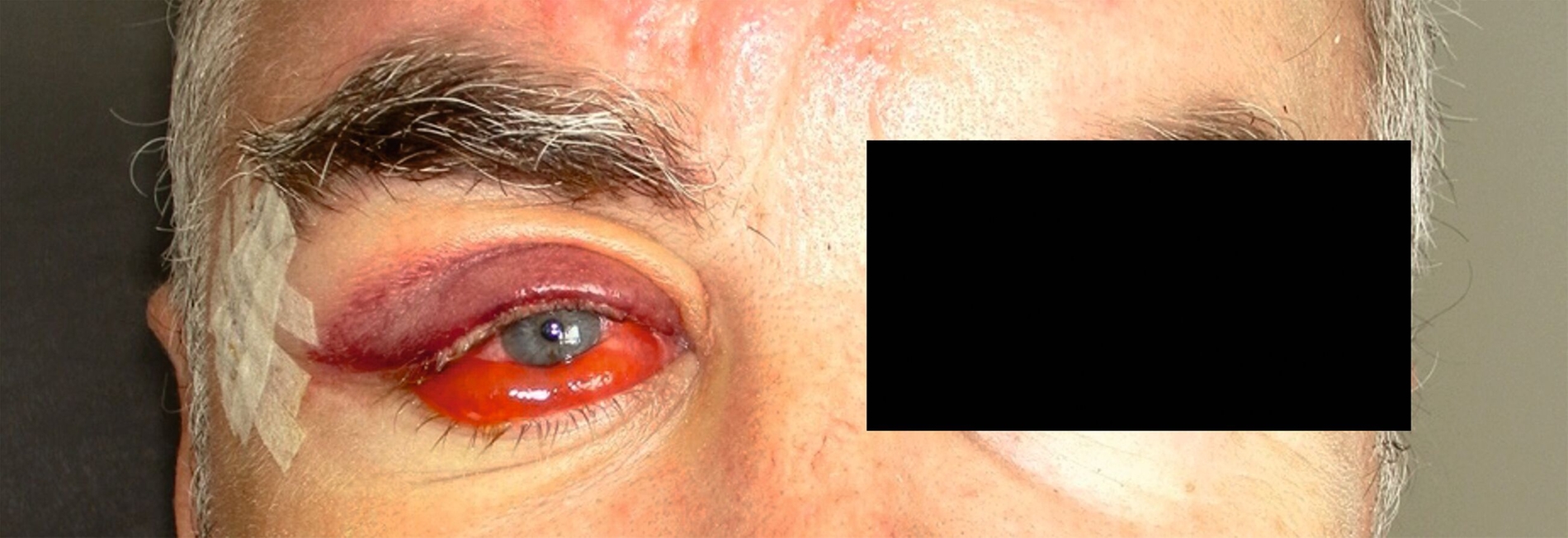

Im Juni 2022 stellte sich ein 56-jähriger Patient nach Überweisung durch die Augenklinik in domo aufgrund einer orbitalen Raumforderung der rechten Seite mit dadurch bedingter Bulbusprotrusion in der Poliklinik der Mund-, Kiefer- und Gesichtschirurgie der Universitätsmedizin Mainz vor. Der Exophthalmus (Abbildung 1) bestehe anamnestisch seit sechs Monaten, weitere klinische Symptome wies der Patient allerdings nicht auf. Insbesondere Visus, Gesichtsfeld und Bulbusmotiliät zeigten sich regelrecht.
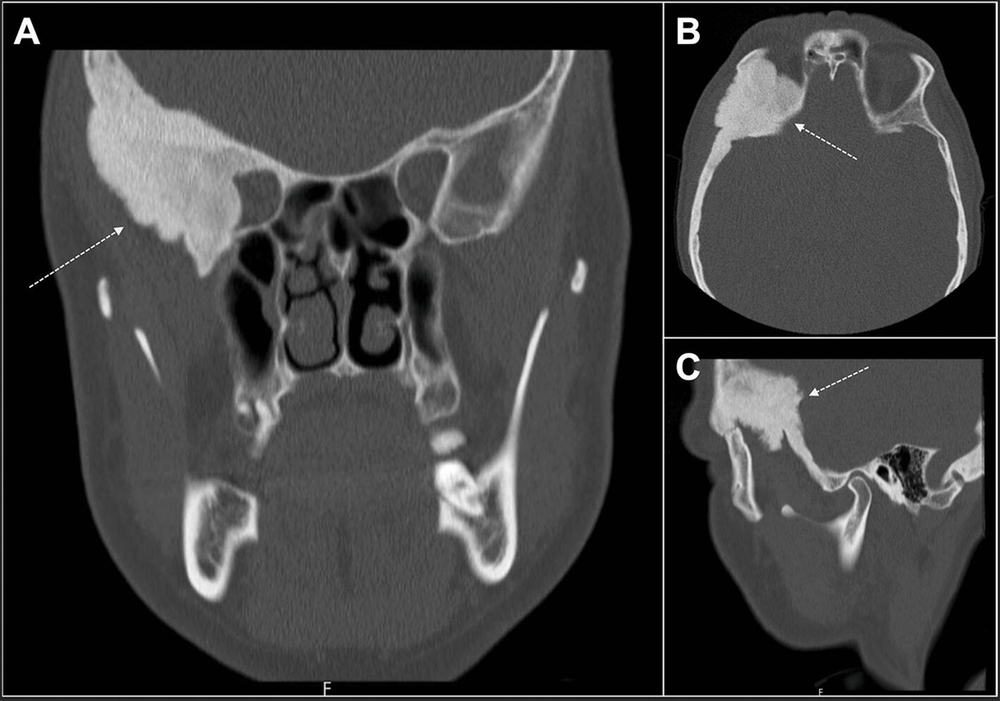
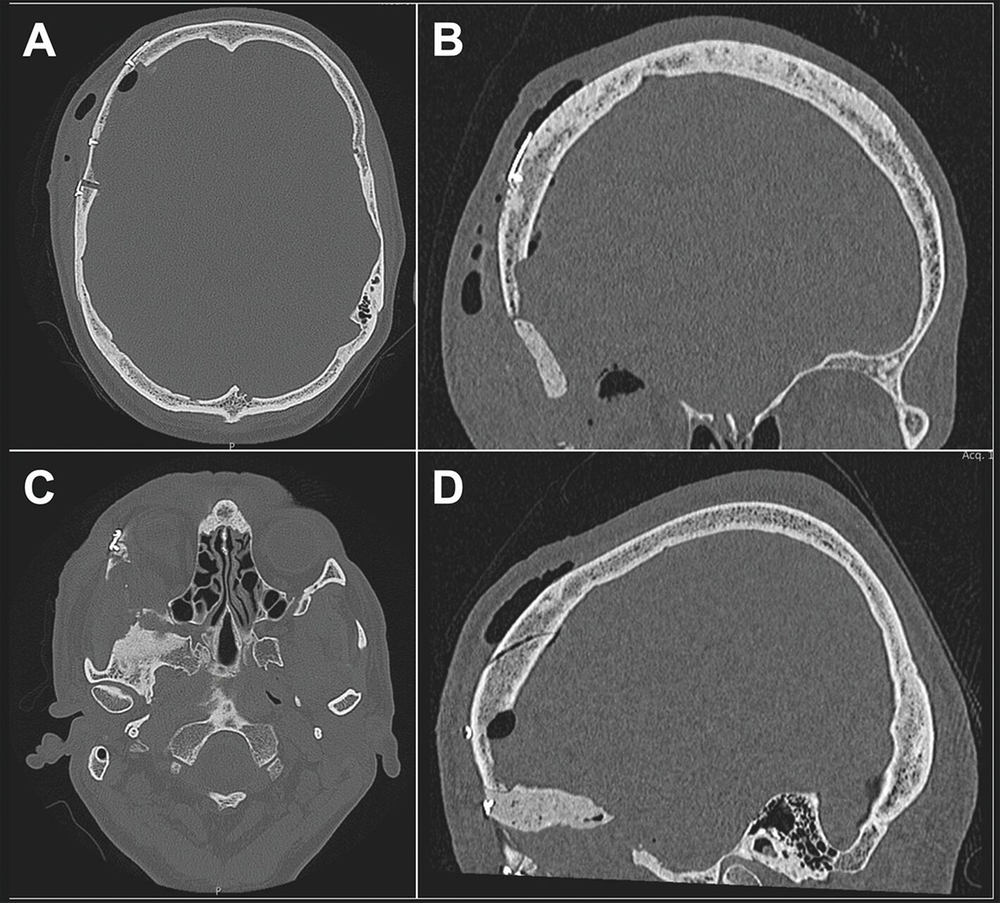
Die weitere Anamnese war unauffällig, es waren keine Vorerkrankungen bekannt und laborchemisch zeigten sich die Entzündungs- und Schilddrüsenparameter normwertig. Radiologisch wurde in einem alio loco durchgeführten CT eine sklerosierte, expansive Raumforderung am Os sphenoidale mit Einengung der Orbita (Abbildungen 2 und 3) ohne sicheren Malignitätsausschluss beschrieben, so dass differenzialdiagnostisch eine fibröse Dysplasie sowie ein lokalisierter Morbus Paget in Betracht gezogen wurden.
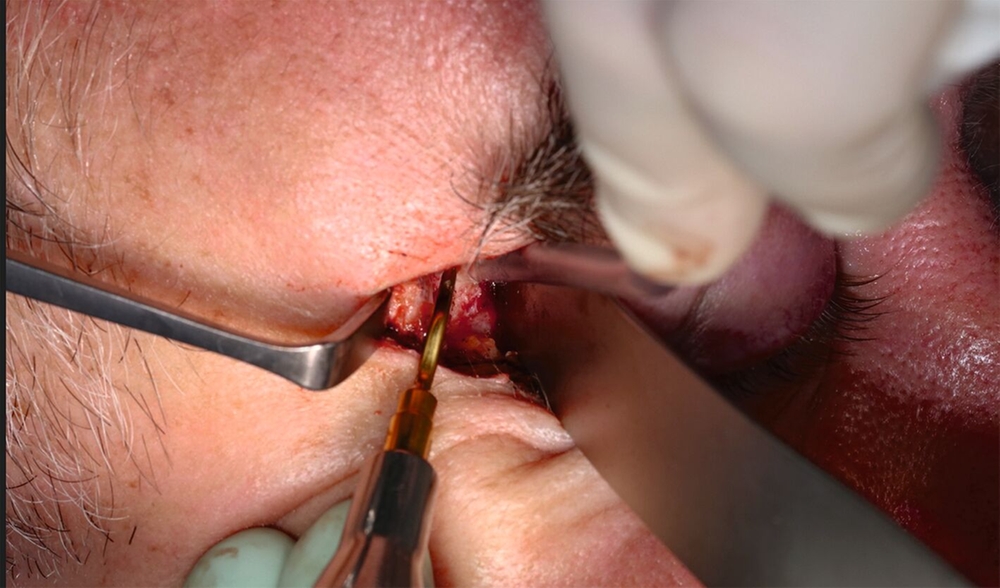
Zur Diagnosesicherung erfolgte bereits am nächsten Tag die offene Probenentnahme in Intubationsnarkose. Nach lateroorbitaler Schnittführung erfolgte die Stückosteotomie des zygomaticofrontalen Übergangs zur Darstellung der knöchern-proliferativ anmutenden Gewebemassen sowie die Probenentnahme durch Osteotomie — sowohl intraorbital als auch in der Fossa temporalis (Abbildung 4). Histopathologisch zeigten sich Lamellenknochenanteile mit Markraumfibrose sowie Proliferaten mit überwiegend monomorphen hyperchromatischen Zellkernen. Ergänzende immunhistochemische Zusatzuntersuchungen zeigten keine Anfärbung mit Antikörpern gegen Panzytokeratin, SM-Aktin, p40, S-100, GFAP, Synaptophysin, Chromogranin A, CD34, ERG, D2-40, CD31 oder WT1. Allerdings wiesen die EMA-Antikörper eine mäßig starke durchgängige Anfärbung und die Antikörper gegen Progesteronrezeptor eine kräftige nukleäre Anfärbung auf. Weiterhin zeigten die Präparate eine kräftige, durchgängige Anfärbung mit Antikörpern gegen SSTR2A. In Zusammenschau dieser Befunde mit dem morphologischen Bild und dem Nachweis eines einzelnen Psammomkörperchens (Verkalkung) bei fehlendem Nachweis von Mitosefiguren ließ sich die Diagnose eines meningothelialen Meningeoms ZNS Grad 1 nach WHO-Klassifikation stellen.
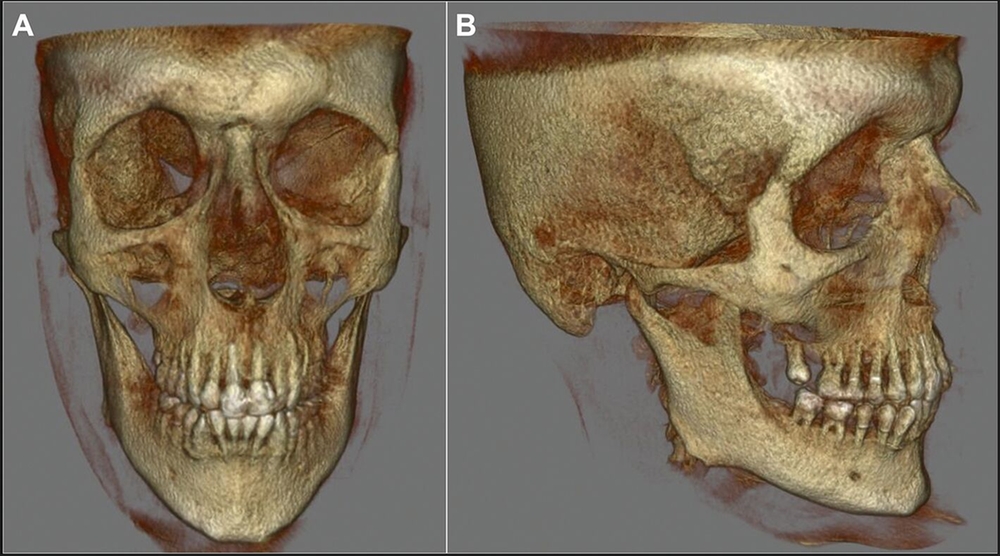
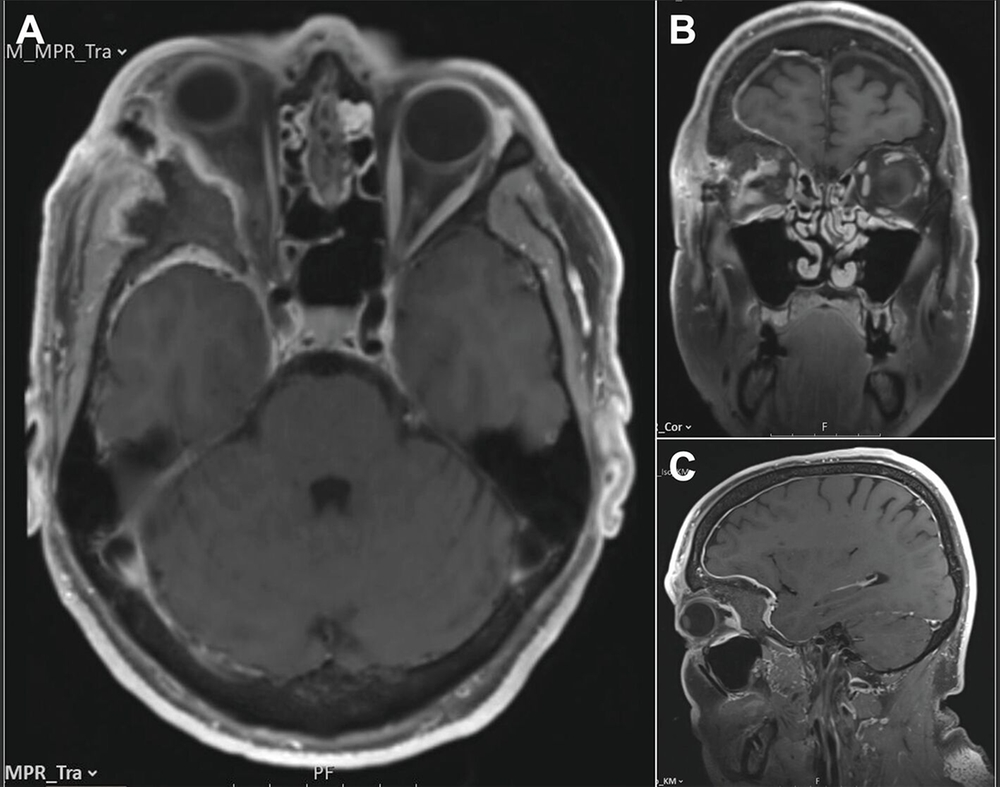
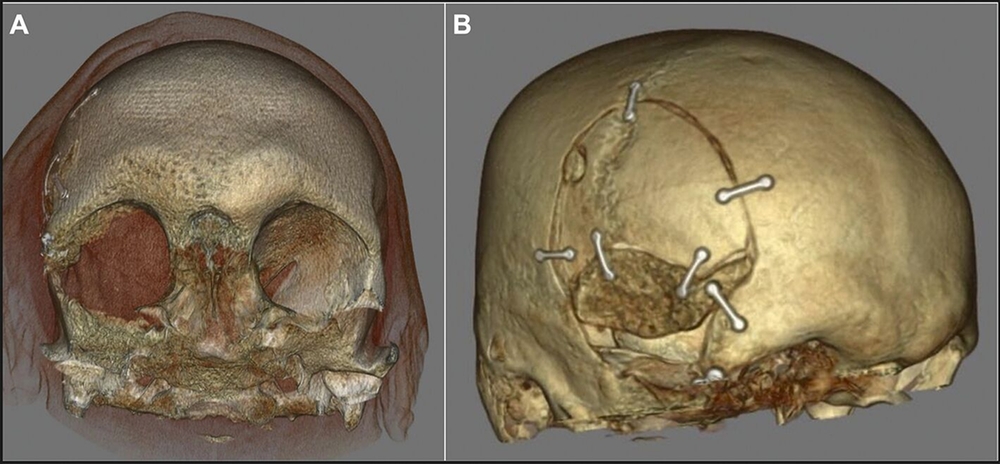
Unter Einbeziehung der Kollegen der Neurochirurgie erfolgte daraufhin die Vorstellung des Patienten im interdisziplinären neuroonkologischen Tumorboard mit Empfehlung zur Resektion des Tumors. Ein präoperativ ergänzend durchgeführtes MRT zeigte ein sich diffus intraossär ausbreitendes Meningeom ausgehend von Os zygomaticum / Os sphenoidale / Os frontale mit Einbeziehung von Orbitadach und lateraler Orbitawand unter Verdrängung des rechten Frontallappens, des rechten Temporallappens und der Pelottierung des rechten Orbitatrichters mit Kompression des Nervus opticus. Neben dem intraossären En-plaque-Wachstum zeigte sich ein nur geringes intradurales rasenartiges Wachstum frontal und temporal (Abbildung 5). Der Tumor konnte infolgedessen neurochirurgisch reseziert werden (Abbildungen 6 und 7), der Patient wurde nach neuropathologischer Diagnosesicherung eines meningothelialen Meningeoms ZNS WHO-Grad 1 mit Infiltration der Dura und des Knochens nach regelrechtem postoperativem Verlauf in die ambulante Nachbetreuung entlassen. Gemäß Tumorboardbeschluss wird eine jährliche klinische Nachsorge nach einem Ausgangs-MRT nach drei Monaten erfolgen.
Diskussion
Obwohl in der Zahnmedizin wie in der MKG-Chirurgie eine große Bandbreite an unterschiedlichen Tumorerkrankungen diagnostiziert und therapiert wird, stellen Tumorerkrankungen des zentralen Nervensystems in unserem Fachgebiet eine Rarität dar. Der dargestellte Fall zeigt allerdings eindrücklich, dass auch diese Erkrankungen differenzialdiagnostisch von Bedeutung sein können.
Bei Tumoren des zentralen Nervensystems handelt es sich um eine sehr heterogene Gruppe verschiedener Tumorentitäten, die anhand der WHO-Klassifikation — basierend auf ihren histologischen, molekularen und immunhistochemischen Eigenschaften — in zwölf Untergruppen eingeteilt werden können: Gliome, ependymale Tumoren, Choroidalplexustumoren, embryonale Tumoren, Pinealistumoren, Tumoren der Hirn- und Spinalnerven, Meningeome, mesenchymale nicht meningotheliale Tumoren, hämatolymphoide Tumore, Keimzelltumore, Tumore der Sella-Region und ZNS-Metastasen. Aktuell befindet sich die WHO-Klassifikation in der fünften Auflage aus dem Jahr 2021, wobei diese nun maßgeblich durch neue Erkenntnisse der Tumorgenomik geprägt ist. Dementsprechend werden auch Mutationen und Genfusionen für die Einteilung in Betracht gezogen [Louis et al., 2021].
Zusätzlich besteht weiterhin eine Graduierung im Rahmen der WHO-Klassifikation, die vor allem im klinischen Alltag große Relevanz aufweist. Nach der alten Klassifikation wurden vier Grade der Hirntumore unterschieden, wobei sich darin vor allem die prognostische Bedeutung widerspiegelt. Die aktuelle Klassifikation verwendet nun statt römischer Ziffern arabische und die Graduierung von Neoplasien findet innerhalb ihres Typs und nicht mehr übergreifend über verschiedene Entitäten statt [Louis et al., 2021].
Meningeome sind meist gutartige und langsam wachsende Tumore, ausgehend von den Deckzellen der Arachnoidea, wodurch sie an jeder intrakraniellen oder spinalen und von Dura bedeckter Oberfläche auftreten können. In seltenen Fällen können sie außerdem intraventrikulär auftreten [Buerki et al., 2018]. Es handelt sich bei Meningeomen um den häufigsten primär intrakraniellen Tumor mit einer altersabhängigen Inzidenz, die mit dem Lebensalter zunimmt und einen Häufigkeitsgipfel im 50. Lebensjahr aufweist [Ostrom et al., 2022]. Gleichzeitig ist die Prävalenz in der afro-amerikanischen Bevölkerung im Vergleich zu Kaukasiern höher. Frauen sind außerdem häufiger betroffen als Männer in einem Verhältnis von 3:2 [Ostrom et al., 2022; Ostrom et al., 2019]. Bisher wurde für die Mehrheit der Fälle keine kausale Ursache identifiziert, jedoch stellt die ionisierende Strahlung, beispielsweise im Rahmen der radiologischen zahnärztlichen Diagnostik, einen erheblichen Risikofaktor dar (sechs- bis zehnfach erhöhtes relatives Risiko), ohne allerdings eine Dosis-Wirkungs-Beziehung aufzuweisen [Wiemels et al., 2010]. Die Latenzzeit ist dabei sehr variabel. Weiterhin ist die Erbkrankheit Neurofibromatose Typ 2 mit einem gesteigerten Auftreten von Meningeomen assoziiert [Rogers et al., 2015].
Als überwiegend benigne Tumore wachsen Meningeome verdrängend und nicht infiltrativ, wobei das Wachstum charakteristischerweise sehr langsam fortschreitet. Verschiedene Studien haben für asymptomatische Meningeome ein Wachstum von 2-4 mm/Jahr nachgewiesen [Buerki et al., 2018; Chamberlain, 2017]. Folglich zeigen sie sich lange Zeit symptomlos und stellen häufig Zufallsbefunde dar. Eine Ausnahme sind Meningeome in der Schwangerschaft, die ein verhältnismäßig schnelles Wachstum aufweisen können, am ehesten bedingt durch Östrogen- oder Progesteronrezeptoren des Tumors. Beim Auftreten von Symptomen sind diese nicht pathognomonisch und in der Regel durch eine allgemeine Hirntumorsymptomatik wie Kopfschmerzen und verdrängungsbedingte Wesensveränderungen, fokal neurologische Defizite oder epileptische Anfälle, bestimmt. In Abhängigkeit von der Lokalisation können Meningeome allerdings auch mit spezifischen Symptomen einhergehen. Als Beispiele sind hier das Mantelkantensyndrom mit Paraparese der Beine und unkontrollierter Blasenentleerung, das Syndrom der Olfaktoriusrinne mit Hyposmie und Parosmie oder das Foster-Kennedy-Syndrom mit kompressionsbedingter ipsilateraler Optikusatrophie und kontralateraler Stauungspapille zu nennen [Buerki et al, 2018].
Davon lässt sich allerdings die Gruppe der sphenoorbitalen Meningeome abgrenzen, die bereits 1938 unter dem Synonym „meningioma en plaque“ beschrieben wurde. Diese Tumoren zeichnen sich durch ein intraossäres En-plaque-Wachstum mit einem rasenartigen intraduralen Befall aus und sind typischerweise in der Orbita lokalisiert. Folglich werden die Patienten in der Regel klinisch durch einen Exophthalmus auffällig. Weitere mögliche Symptome sind Visusminderungen, Gesichtsfeldeinschränkungen, Doppelbilder, trigeminale Hypästhesien oder retroorbitale Schmerzen [Ringel et al., 2007].
Aufgrund charakteristischer bildmorphologischer Eigenschaften steht diagnostisch die Bildgebung mittels CT oder MRT an erster Stelle. Im CT präsentiert sich das Meningeom meist leicht hyperintens mit einer seitlichen Duraverdickung, dem sogenannten „Dural Tail Sign“. Zusätzlich weisen Meningeome ein ausgeprägtes Kontrastmittel-Enhancement bei variabler Ödemneigung auf, wodurch sich das Bild eines Schneeballs ergibt. Gelegentlich sind außerdem Verkalkungen nachweisbar. Im MRT zeigen sich Meningeome in der T1-Wichtung isointens und in der T2-Wichtung hyperintens [Buerki et al., 2018].
Da die Diagnosestellung meist bereits anhand der Bildgebung möglich ist, ist die Durchführung einer Probebiopsie nur in Ausnahmefällen notwendig. Histopathologisch findet man mesenchymales Ursprungsgewebe mit einer charakteristischen Zwiebelschalenformation der Tumorzellen und einer Hypervaskularisierung. Verkalkungen stellen sich mikroskopisch als sogenannte Psammomkörper dar [Buerki et al., 2018]. Zur immunhistochemischen Diagnosesicherung eignet sich die Anfärbung mit Antikörpern gegen Somatostatin Rezeptor 2A [Menke et al., 2015]. Außerdem weisen 70 bis 80 Prozent der Meningeome eine Progesteronrezeptor-Expression auf, wodurch sich das schnellere Wachstum während der Schwangerschaft erklären lässt [Hsu et al., 1997]. Weiterhin sind bisher zahlreiche Mutationen identifiziert worden, die mit verschiedenen Eigenschaften korrelieren und insbesondere das epigenetische Methylierungsmuster steht in Zusammenhang mit der Rezidiv-Wahrscheinlichkeit [Olar et al., 2017]. Das Meningeom kann auf Grundlage der Mitoserate sowie der Invasivität den WHO-Graden I-III zugeordnet werden. Den überwiegenden Anteil (81,1 Prozent) stellen typische Meningeome Grad I, während es sich bei 16,9 Prozent um atypische Meningeome Grad II mit höherer Mitoserate und invasiverem Wachstum handelt. Nur wenige Meningeome (1,7 Prozent) werden als anaplastisch Grad III zugeordnet [Ostrom et al., 2019; Ostrom et al., 2016].
Die Überlebensrate bei Meningeomen ist maßgeblich vom WHO-Grad und der möglichen Therapie abhängig, kann aber im Allgemeinen als sehr hoch angesehen werden. Insgesamt liegt die Zehn-Jahres-Überlebensrate bei 57,1 Prozent und für jüngere Patienten zwischen 20 und 44 Jahren bei Diagnosestellung sogar bei 77,7 Prozent. Mit zunehmender Aggressivität des Tumors und höheren Rezidivraten sinkt gleichzeitig die Lebenserwartung. Meningeome des WHO-Grades III zeigen dabei — trotz radikaler Therapieversuche — eine Zehn-Jahres-Überlebensrate von 0 Prozent [Ostrom et al., 2016]. Therapeutisch besteht die Möglichkeit einer „watch and wait“-Strategie, die insbesondere bei älteren Patienten und fehlenden Symptomen in Betracht gezogen werden sollte. In den meisten Fällen ist allerdings eine radikale chirurgische Therapie möglich, die aus der vollständigen Exstirpation des Tumors mit der duralen Ansatzstelle besteht. Diese operative Tumorresektion stellt bei Meningeomen die primäre Therapieform dar [Goldbrunner et al., 2019]. Unterstützend kann gerade bei stark vaskularisierten Tumoren in seltenen Fällen eine präoperative Embolisation in Betracht gezogen werden. Bei Inoperabilität eines Meningeoms, beispielsweise durch die Beteiligung venöser Sinus, Arterien, Hirninfiltration oder signifikanter Komorbiditäten, kann die Möglichkeit einer Strahlentherapie diskutiert werden [Buerki et al., 2018; Rogers et al., 2015].
")
