Raumforderung des Kiefers führt zur Erstdiagnose des Noonan-Syndroms
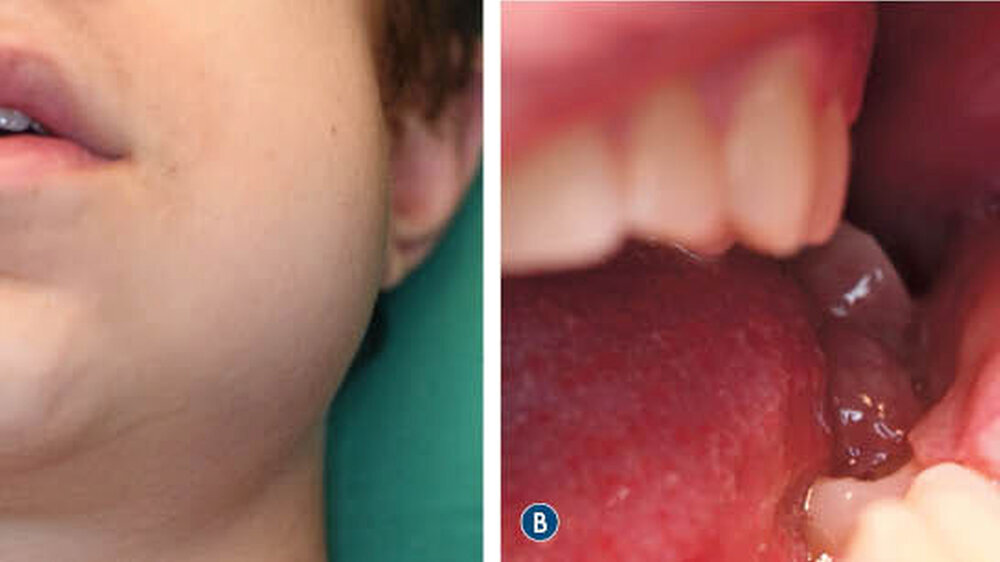

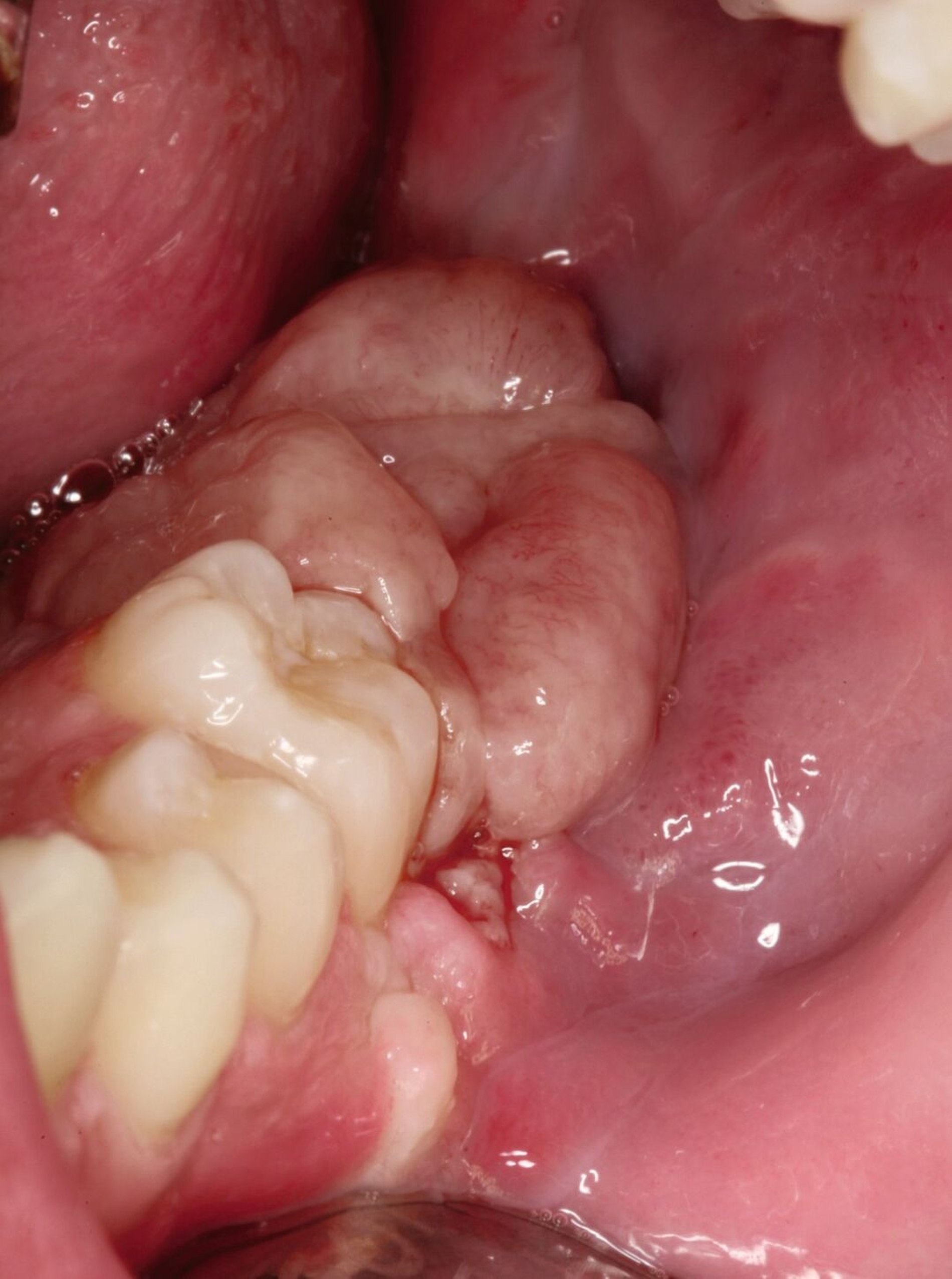
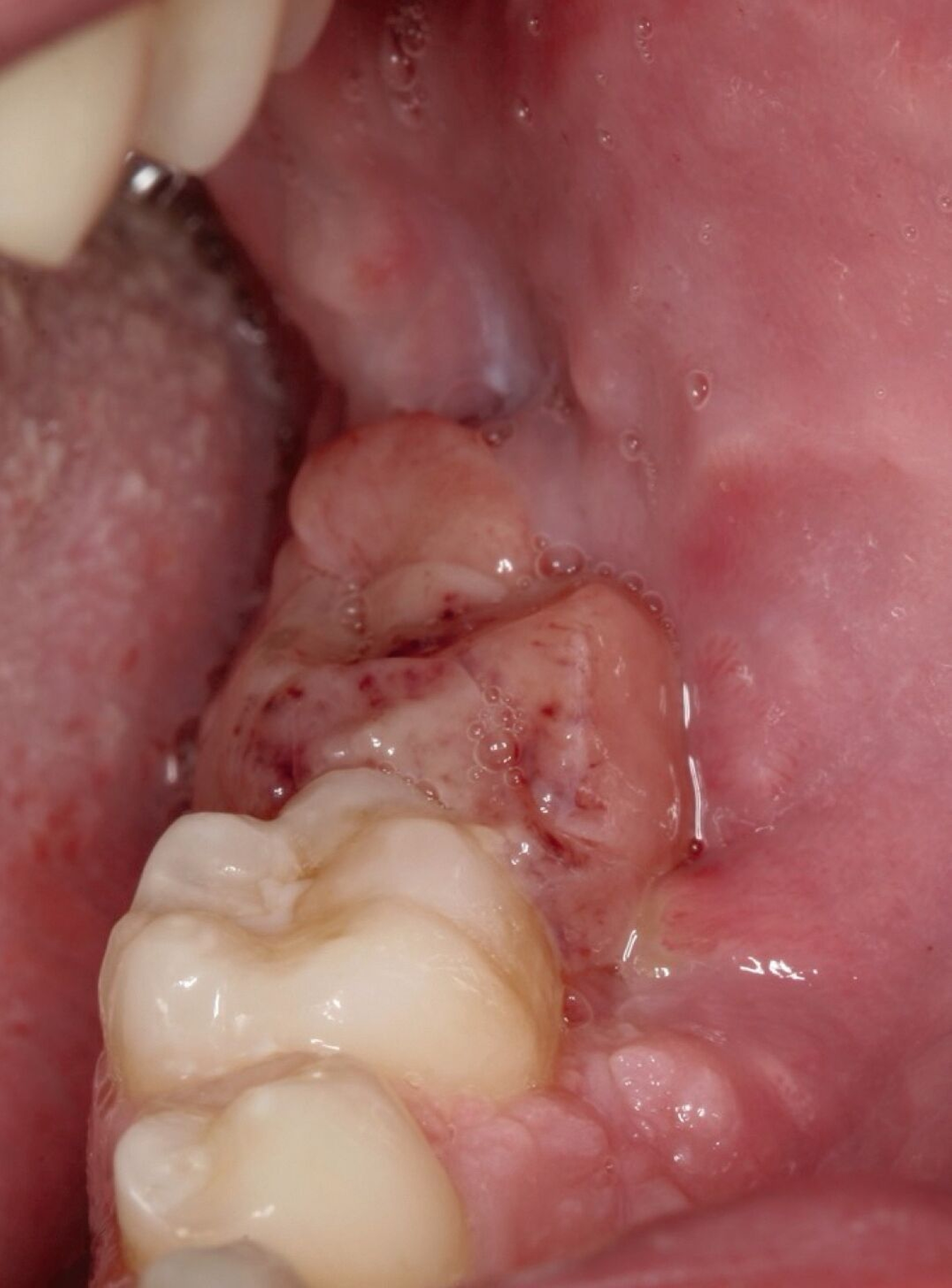
Ein 16-jähriger Junge stellte sich nach Überweisung durch seine behandelnde Kieferorthopädin aufgrund einer progredienten Schwellung des linken Unterkiefers in der Klinik und Poliklinik für Mund-, Kiefer- und Gesichtschirurgie vor. In der klinischen Untersuchung zeigte sich ein prominenter Befund distal der Zahnreihe mit hyperplastisch wirkender Schleimhaut (Abbildung 1).
Im Alter von drei Jahren war wegen einer Teilleistungsstörung, einer Pulmonalstenose und den augenärztlichen Befunden eines Hypertelorismus, eines Epicanthuis medialis beidseits, einer Ptosis des rechten Auges und eines dekompensierten Strabismus divergens bereits eine genetische Testung des Patienten durchgeführt worden. Jedoch hatte diese weder für das DiGeorge-Syndrom noch für das Wiliam-Beuren-Syndrom einen positiven Befund ergeben. Die Familienanamnese zeigte bezüglich genetischer Erkrankungen keine Auffälligkeiten, ebenso wie die laborchemische Auswertung des Blutes.
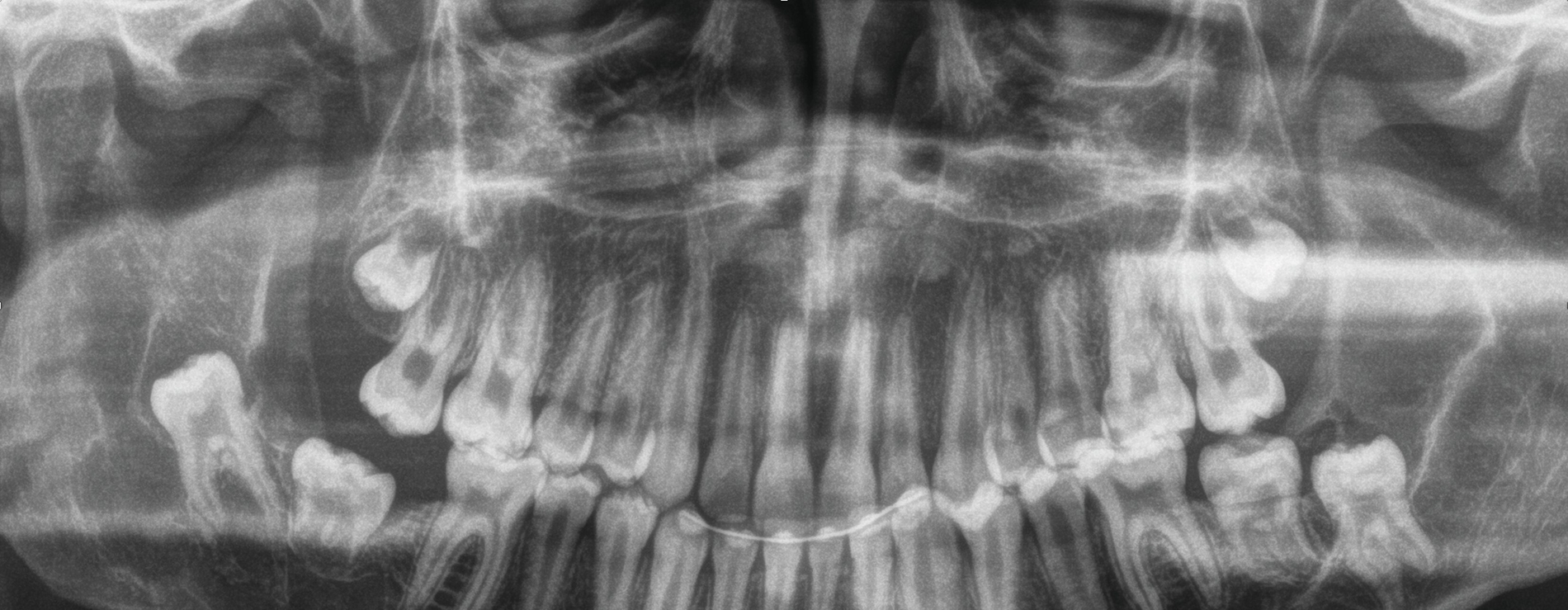
Zur weiteren Diagnostik wurde eine Panoramaschichtaufnahme durchgeführt. In dieser war ein radioluzenter, mehrkammeriger, scharf begrenzter Befund im Bereich des linken Unterkiefers um den retinierten Zahn 38 darstellbar. Auch auf der Gegenseite war eine, hier eher unklar begrenzte, Radioluzenz um den retinierten und verlagerten Zahn 48 zu erkennen (Abbildung 2).
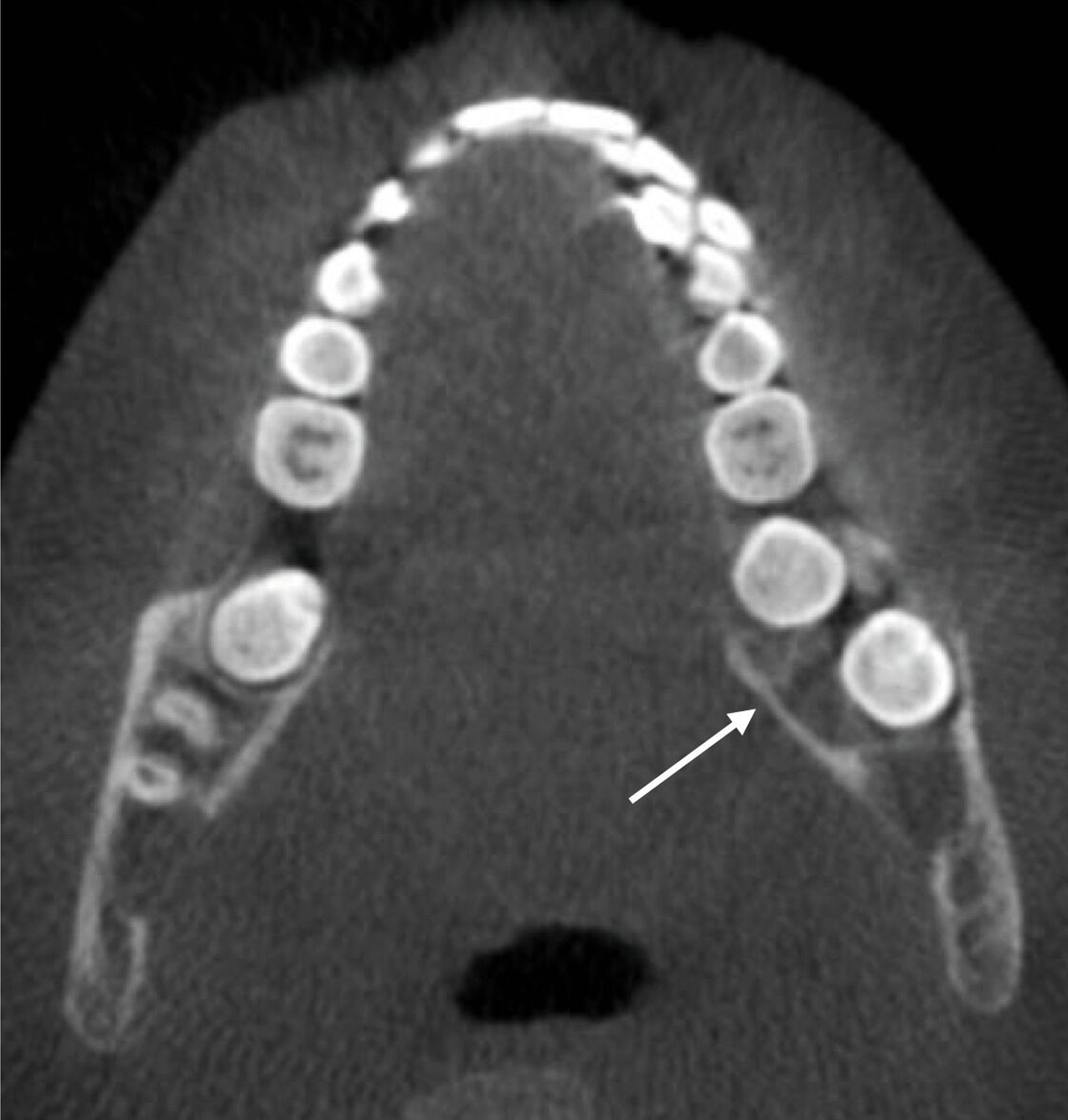
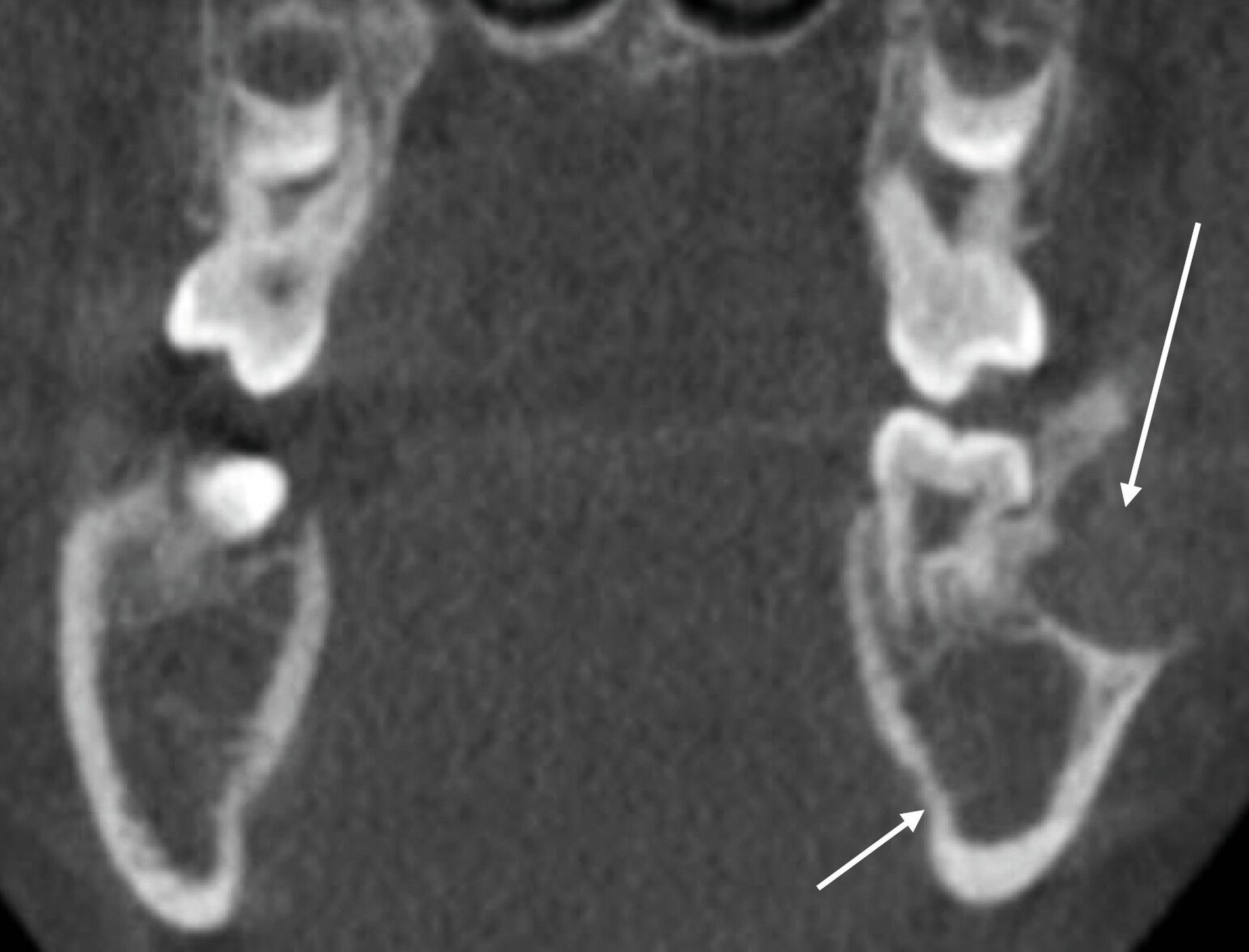
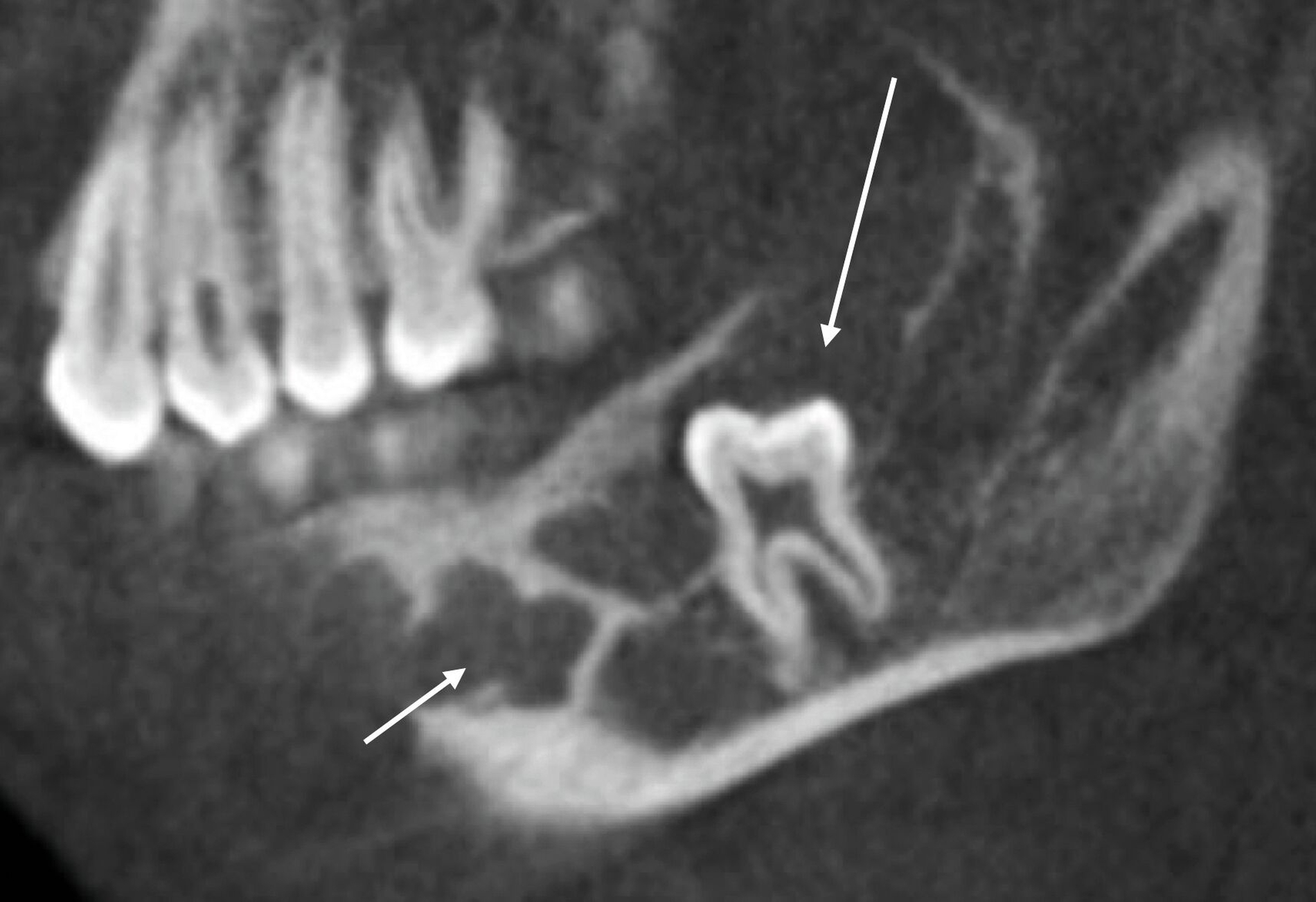
Um diese Befunde bezüglich ihrer Lage und ihrer Ausdehnung genauer zu charakterisieren, erfolgte anschließend eine dreidimensionale Aufnahme in Form einer Digitalen Volumentomografie. In dieser ließen sich analog zu den Befunden in der zweidimensionalen Bildgebung gelappte hypodense Raumforderungen darstellen. Der sklerotische Randsaum und die – im Vergleich zur Spongiosa – hypodensere Struktur des Befunds im dritten Quadranten waren klar zu erkennen (Abbildung 3).
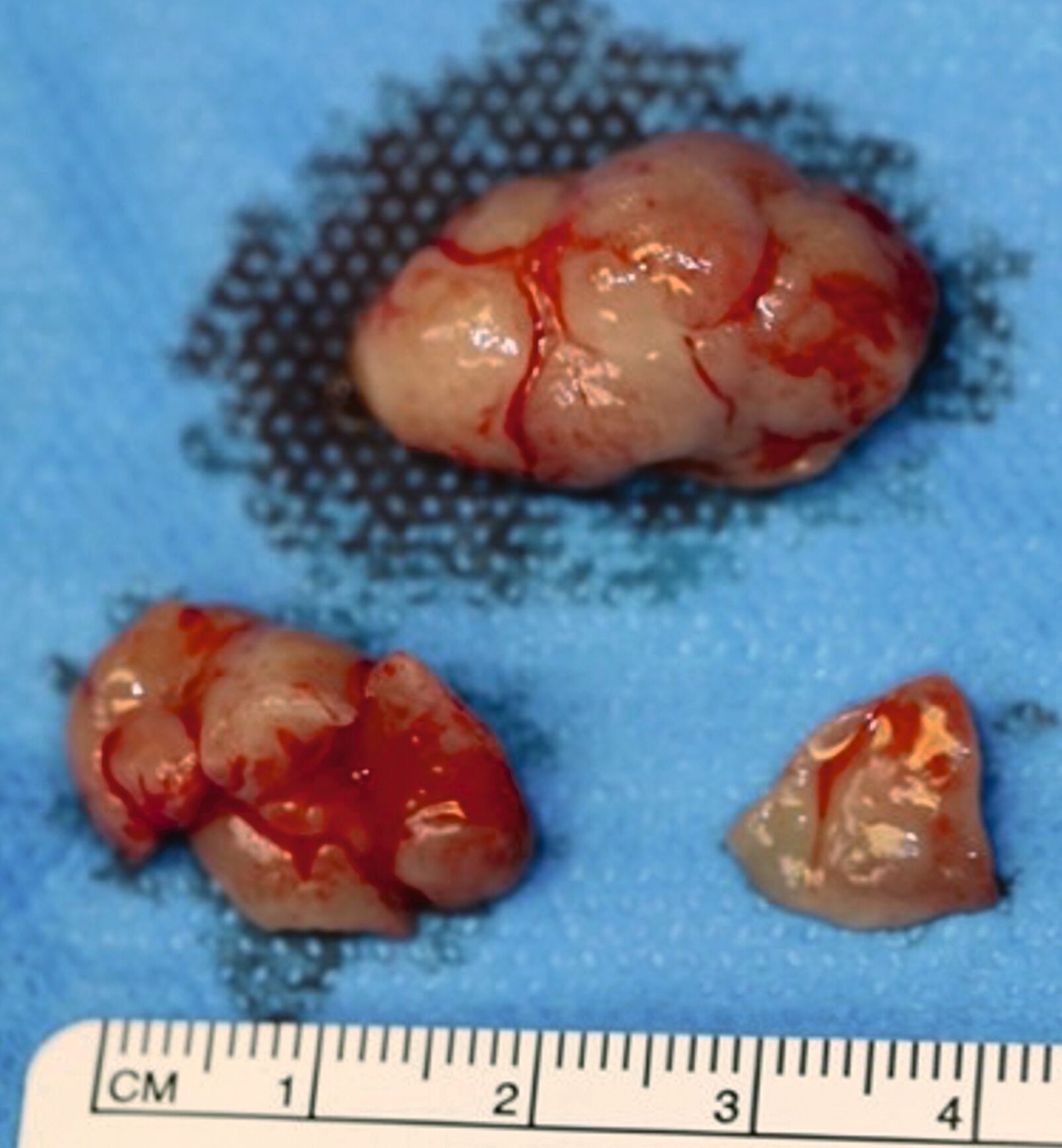
Bei Verdacht auf das Vorliegen einer Keratozyste und zum Ausschluss eines Malignoms wurde die chirurgische Entfernung der Befunde in Intubationsnarkose durchgeführt. Hierbei erfolgte die Zystektomie beider Raumforderungen gemeinsam mit der Osteotomie der Zähne 37, 38, 47 und 48. Insbesondere in regio 38 zeigte der Befund eine direkte Kontinuität zwischen Knochen und Weichgewebe. Das Resektat wies eine gelbliche Farbe und eher weiche Konsistenz auf (Abbildung 4).
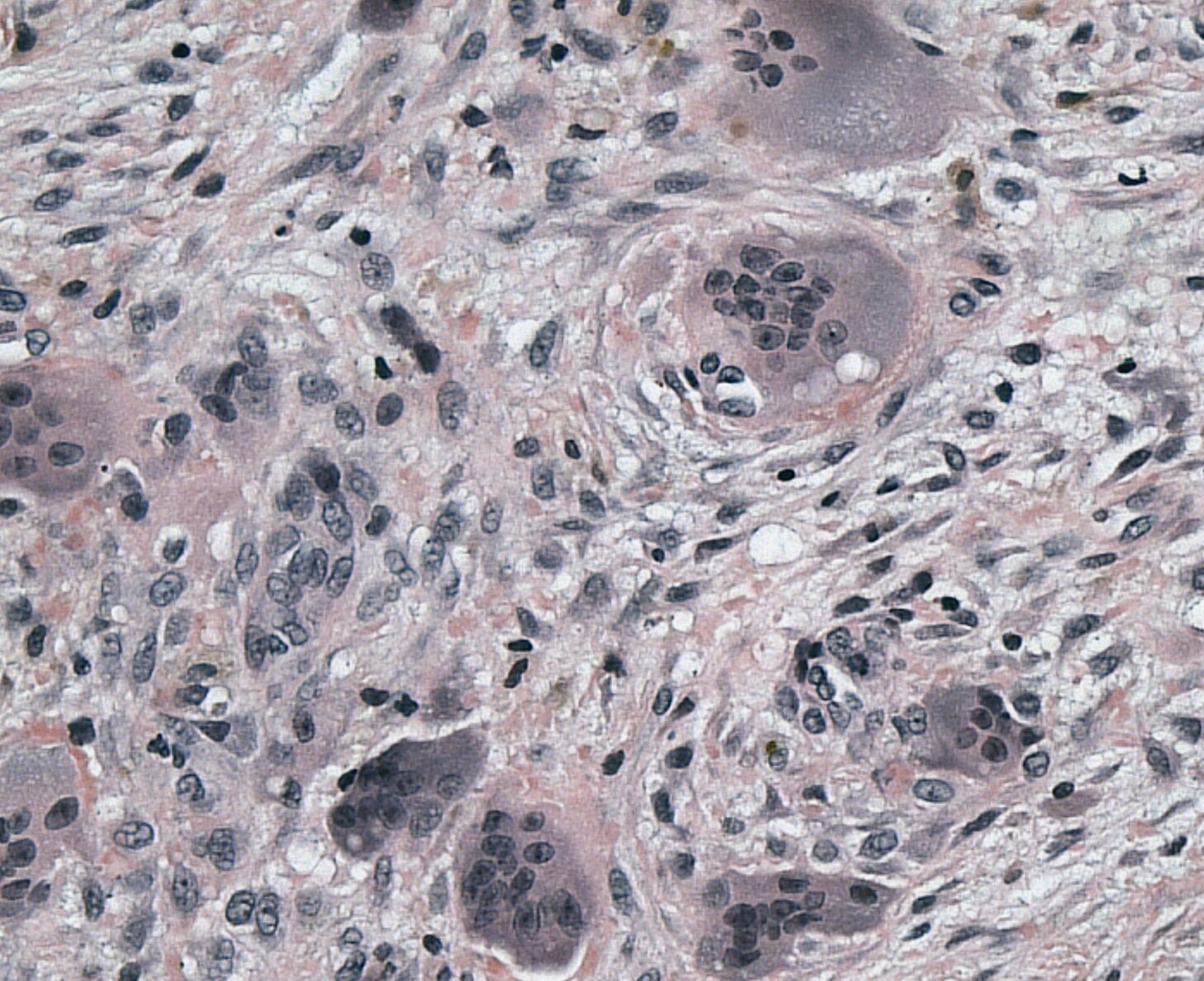
In Anbetracht der doch beträchtlichen Größe der Wundhöhle wurde diese anschließend mit Beckenkammspongiosa aufgefüllt. In der anschließenden histopathologischen Aufarbeitung kam eine fragmentierte spindelzellige und riesenzellhaltige Läsion des Weichgewebes mit lokal den Knochen destruierendem Wachstumsverhalten zur Darstellung. Zudem zeigte der Befund starke Entzündungszeichen, was die differenzialdiagnostische Beurteilung deutlich erschwerte. Zur Klärung der Dignität des Befunds wurde dieser zur referenzpathologischen Mitbeurteilung versandt. Auch in dieser wurde ein polypoides, teils plattenepithelial überkleidetes Exzidat mit ausgedehnter Ulzeration sowie einem kapillarreichen Granulationsgewebe mit diffus verteilten Riesenzellen beschrieben (Abbildung 5). In beiden pathologischen Berichten wurde die Raumforderung als primär mit einem Riesenzellgranulom vereinbar erachtet.
In der engmaschigen klinischen Nachsorge zeigte sich bei zunächst blandem Heilungsverlauf nach einer Latenz von drei Monaten schließlich ein Rezidiv im linken Unterkiefer. In Anbetracht dessen und bei Vorliegen eines auffälligen Habitus (vor allem eines dezenten Hypertelorismus) des Patienten erfolgte die gemeinsame Fallbesprechung mit den Kollegen der Kinderonkologie.
In Anbetracht des aktuell neu aufgetretenen Befunds eines Riesenzellgranuloms fand eine erneute genetische Testung statt, die diesmal eine heterozygote SOS1-Mutation in Exon 11 (c.1654A>G) ergab. Hiermit wurde schließlich die Erstdiagnose eines Noonan-Syndroms gestellt.
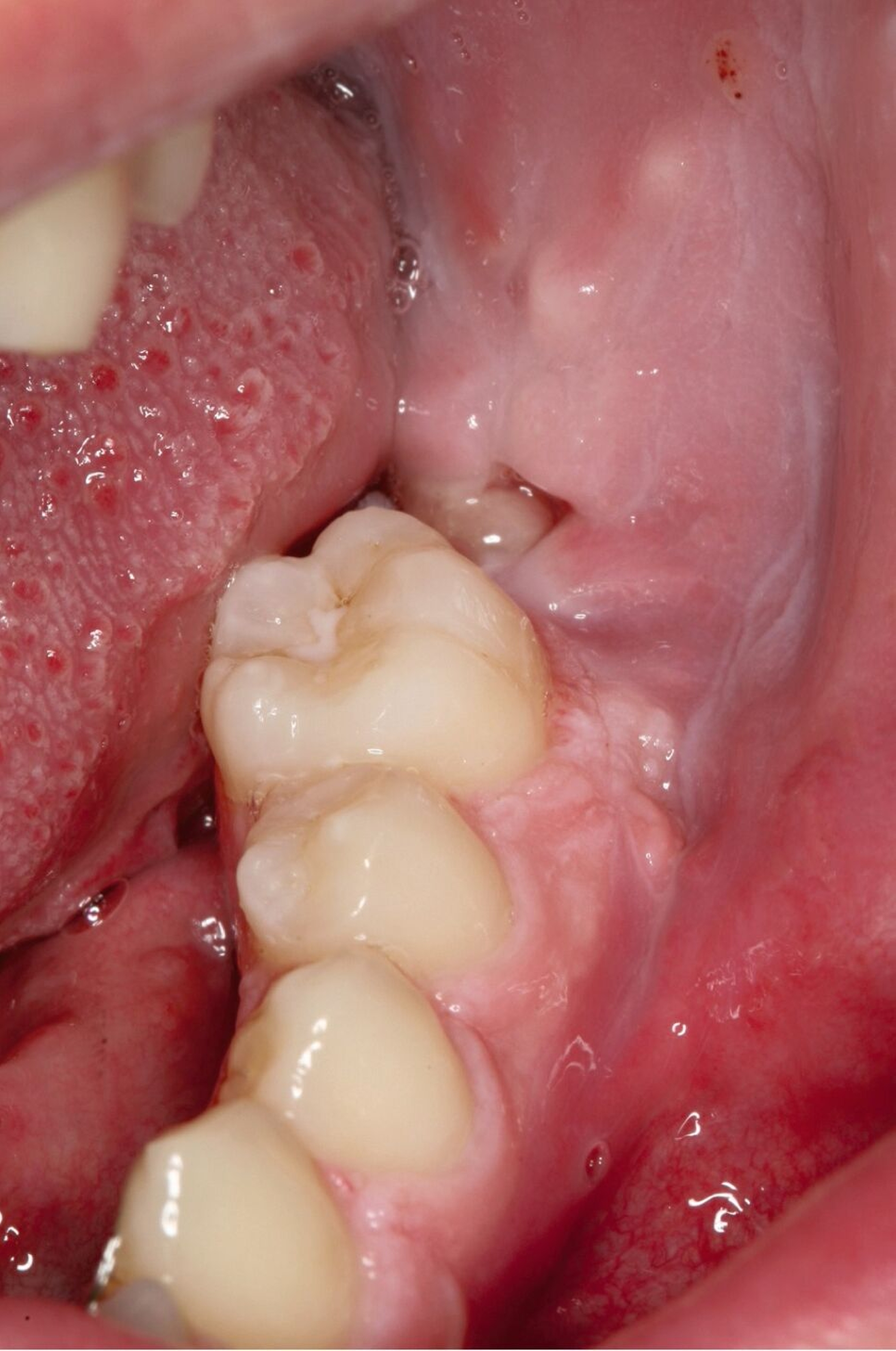
Aufgrund des nach der primären Resektion erneut aufgetretenen Rezidivs, des jungen Alters des Patienten und der zu antizipierenden Morbidität bei vollständiger weiterer Exzision wurde die gemeinsame Entscheidung zur Lokaltherapie mit Steroiden getroffen. Diese erfolgte durch wöchentliche Injektion mit Triamcinolon über einen Zeitraum von zehn Wochen. Nach einer Therapiepause von drei Monaten zeigte sich ein erneutes Rezidiv, das jedoch durch die wöchentliche Injektion des Steroids vollständig und dauerhaft therapiert werden konnte. In Abbildung 6 ist der Heilungsverlauf unter regelmäßiger Injektion des Kortikosteroids Triamcinolon über einen Zeitraum von drei Monaten dargestellt. Eine weitere Therapie zeigte sich auch im klinischen Verlauf von mehreren Jahren als nicht notwendig.
Diskussion
RASopathien sind eine bekannte Gruppe von Fehlbildungssyndromen, die durch Keimbahnmutationen verursacht werden, die für Komponenten des RAS/MAPK-Signalwegs kodieren. Sie umfassen das Noonan-Syndrom, Neurofibromatose Typ 1, das Costello- und das LEOPARD-Syndrom sowie weitere sehr seltene Syndrome [Hebron et al., 2022]. Zu den möglichen Symptomen gehören angeborene Herzfehler, Skelettanomalien und andere äußere Merkmale, eine Neigung zu Tumoren und neurokognitive Defizite. Die betroffenen Patienten haben häufig eine erhebliche Beeinträchtigung ihrer Lebensqualität und -dauer [Hebron et al., 2022].
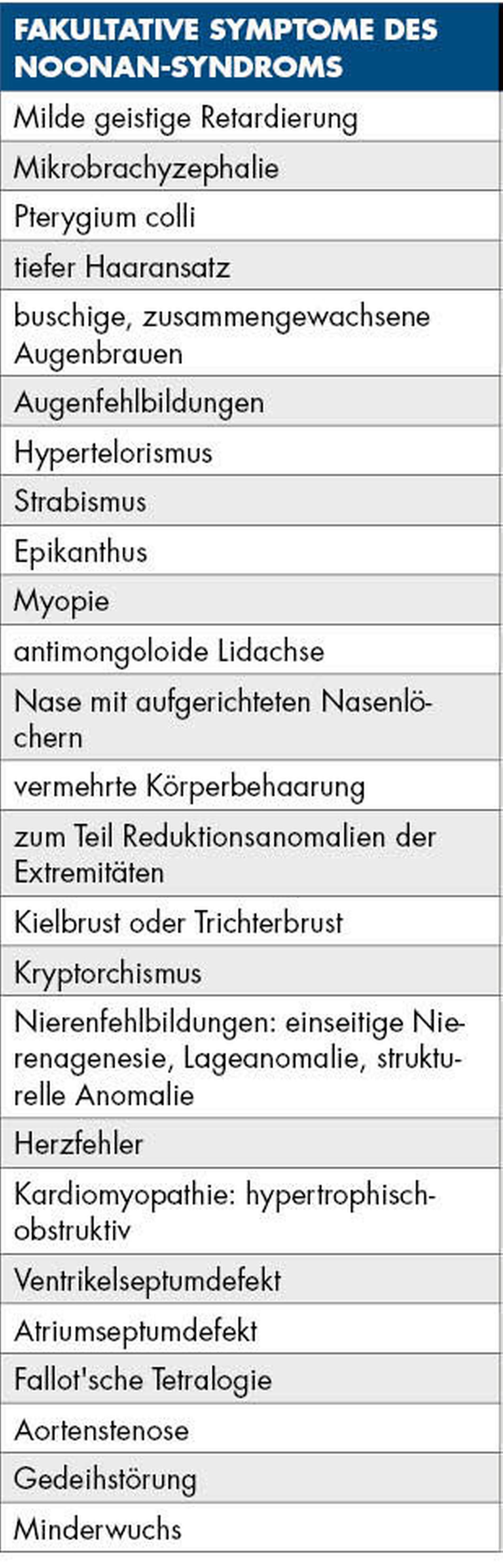
Beim Noonan-Syndrom handelt es sich um einen Fehlbildungskomplex, der in seinem Erscheinungsbild dem Ulrich-Turner-Syndrom ähnlich ist. Mit einer Inzidenz von 1:1.000 Geburten ist das Noonan-Syndrom eines der häufigsten genetischen Syndrome und die zweithäufigste Ursache für angeborene Herzstörungen [Roberts et al., 2013]. Es handelt sich um eine meist autosomal-dominante Anomalie des RAS-MAPK-Signalwegs. Dieser bildet eine zentrale Schnittstelle, über die extrazelluläre Liganden wie Wachstumshormone und Zytokine die Zellproliferation, -differenzierung, und den Zellmetabolismus steuern [Roberts et al., 2013]. Typische Symptome sind – wie im beschriebenen Fall – Hypertelorismus und Ptosis sowie große und tiefsitzende Ohren. Patienten mit einem Noonan-Syndrom weisen häufig einen Kleinwuchs und eine Pulmonalstenose auf. Weitere Symptome, die in ihrer Häufigkeit variieren und nur fakultativ auftreten, sind in Tabelle 1 zusammengefasst [van der Burgt, 2007].
Die Diagnostik gestaltet sich aufgrund der heterogenen klinischen Zeichen und der Ähnlichkeit zu anderen Syndromen schwierig. Liegt der Verdacht auf das Vorliegen eines Noonan-Syndroms vor, so ist die genetische Testung das Mittel der Wahl. Dabei besteht die Möglichkeit einer simultanen Testung multipler Gene und damit auch der Charakterisierung der ursächlichen Genmutation. Je nach zugrundeliegendem Gendefekt liegen unterschiedliche Varianten des Phänotyps vor. Im Vergleich zur Mutation des Gens PTPN11, das als für die Hälfte aller Fälle mit Noonan-Syndrom ursächlich erachtet wird, zeigen Patienten mit einer SOS-1-Mutation deutlich mehr ektodermale Abnormalitäten und weniger kognitive Einschränkungen [Roberts et al., 2013; Zenker et al., 2022]. Zu den möglichen Differenzialdiagnosen zählen insbesondere Syndrome mit ähnlichem Phänotyp wie das Ulrich-Turner-Syndrom, aber auch andere RASopathien wie das Costello-Syndrom, Neurofibromatose 1 und das LEOPARD-Syndrom [van der Burgt, 2007]. Die Therapie der Erkrankung ist primär auf die chirurgische und internistische Behandlung der multiplen Anomalien sowie auf die Unterstützung der individuellen kognitiven Entwicklung ausgerichtet [Zenker et al., 2022]. Zahlreiche Riesenzellläsionen, die durch eine Mutation der Gene PTPN11 oder – wie im vorliegenden Fall – SOS-1 entstehen, sind mit dem Noonan-Syndrom assoziiert.
Riesenzellgranulome sind gutartige, tumorähnliche Läsionen, die am häufigsten den Kiefer betreffen. Das typische periphere Riesenzellgranulom erscheint am Zahnfleischrand als dunkelrote bis bläuliche Epulis. Dabei handelt es sich um eine nicht-neoplastische reaktive Zellproliferation. Klinisch ähnelt der Befund einem pyogenen Granulom. Das periphere Riesenzellgranulom tritt bevorzugt im Seitenzahnbereich auf und ist durch ein hohes Rezidivrisiko charakterisiert. Die endgültige Therapie ist häufig erst durch die Entfernung der Gingiva, des Periosts und des benachbarten Zahnes möglich [Kämmerer und Kunkel, 2008; Gundlach, 2022]. Klinisch ist nicht immer sicher zwischen dem peripheren und dem zentralen Riesenzellgranulom zu unterscheiden. Während das periphere Riesenzellgranulom jedoch eine Erkrankung des Alters darstellt, tritt das zentrale Riesenzellgranulom bevorzugt bei Personen zwischen dem 10. und dem 25. Lebensjahr auf. Primär im Unterkiefer lokalisiert, kann das zentrale Riesenzellgranulom zu Auftreibungen des Knochens und zu Zahnlockerungen führen [Schwarting et al., 2020]. Hier wird häufig zwischen einer nicht-aggressiven und einer aggressiven Form der Erkrankung unterschieden. Die eher seltener (20 bis 30 Prozent) auftretende aggressive Verlaufsform zeichnet sich durch ein schnelles Wachstum mit Durchbrechen der Kortikalis, Wurzelresorptionen, Schmerzen und eine hohe Rezidivneigung aus [EI-Naggar et al., 2017; Gundlach, 2022].
In der radiologischen Bildgebung findet sich – wie im vorgestellten Fall – ein unscharf begrenzter, teilweise polyzystischer Befund, der differenzialdiagnostisch von einem Ameloblastom und der Keratozyste unterschieden werden muss. Makroskopisch zeigt der Befund ein fleischfarbenes, rötlich-braunes, hämorrhagisches Erscheinungsbild. Der histopathologische Befund ist gekennzeichnet durch eine Proliferation mononukleärer Spindelzellen und mehrkerniger Riesenzellen. Die Raumforderung kann lobuliert und von Osteoid und Geflechtknochen umgeben sein [Gundlach, 2022]. Radiologisch und histologisch ist die Differenzialdiagnostik zu anderen riesenzellhaltigen tumorähnlichen und tumorösen Kieferveränderungen nicht immer einfach.
In der Mund-Kiefer-Gesichtschirurgie werden dieser Gruppe folgende Pathologien zugeordnet: das zentrale Riesenzellgranulom, der Riesenzelltumor, der sogenannte braune Tumor bei Hyperparathyreoidismus, der Cherubismus und die aneurysmatische Knochenzyste. Die Differenzialdiagnostik zur aneurysmatischen Knochenzyste stellt sich insbesondere bei längerem Bestehen der Läsion schwierig dar. Während zu Beginn ein ballonartig-zystisches Aussehen für die aneurysmatische Knochenzyste typisch ist, kann diese später als seifenblasenartiger Befund imponieren. Sowohl die charakteristische Lokalisation im Unterkieferseitenzahngebiet, das schnelle Wachstum mit kollateralen Zahnverlagerungen bei erhaltender Vitalität als auch das Auftreten vor dem 30. Lebensjahr erschweren hier die Unterscheidung [EI-Naggar et al., 2017].
Während der echte Riesenzelltumor des Kiefers eine absolute Rarität ist und meist im Bereich der Extremitäten auftritt, stellt der braune Tumor eine weder histologisch noch radiologisch vom zentralen Riesenzellgranulom unterscheidbare Entität dar. Lediglich das Auftreten im höheren Lebensalter und die pathognomonische Erhöhung des Kalzium- und Parathormonspiegels im Blut erlauben hier eine Differenzierung. Das von den Nebenschilddrüsen produzierte Parathormon steuert die Absorption von Kalzium im Darm, die Kalziumrückresorption und die Phosphatexkretion in der Niere und so die osteoklastäre Knochenresorption über den Kalziumhaushalt. Durch die gesteigerte Osteoklastenaktivität und eine vermehrte Knochenresorption mit fibrösem Gewebeersatz kommt es zur Einblutung und dem typischen Bild des braunen Tumors [Gundlach, 2022].
Die Therapie der Wahl für das zentrale Riesenzellgranulom besteht in aller Regel in der Kürettage des Befunds, was jedoch Studien zufolge in elf bis 49 Prozent der Fälle mit einem Rezidiv einhergeht [de Lange et al., 2007]. Als alternative Therapieform zur Limitierung des Resektionsausmaßes kann die Gabe von Steroiden, Calcitonin, Interferon oder Denosumab erwogen werden [EI-Naggar et al., 2017]. Trotz der offensichtlichen Vielfalt der Behandlungsmöglichkeiten existieren bislang keine spezifischen Vorgaben zur Therapie solcher Läsionen bei Patienten mit Noonan-Syndrom [Bufalino et al., 2010]. Dies stellt insbesondere bei Vorliegen multipler oder ausgedehnter Befunde ein Problem dar. Hier muss zwischen Rezidivrisiko beziehungsweise Therapieversagen und der operationsbedingten Morbidität abgewogen werden. 1988 berichteten Jacoway et al. erstmals von der Behandlung von Patienten mit einem zentralen Riesenzellgranulom mittels Kortikosteroiden [Jacoway et al., 1988]. 1994 erschien der erste Fallbericht zu dieser Behandlungsform. Dort führte eine wöchentliche Injektion von Steroiden in die Läsion über einen Zeitraum von sechs Wochen bei drei Patienten zu einer vollständigen Rückbildung [Terry und Jacoway, 1994]. Bei solitären Knochenläsionen durch eine Langerhanszell-Histiozytose ist dies gängige Praxis, auch wenn dazu keine prospektiven kontrollierten Studien vorliegen [de Lange et al., 2007].
Die Wirkungsweise der Kortikosteroide in der Therapie des zentralen Riesenzellgranuloms ist nicht vollständig geklärt. In vitro konnte gezeigt werden, dass Dexamethason einerseits die Proliferation und die Differenzierung von Osteoklastenvorläuferzellen stimuliert, andererseits aber die lakunäre Resorption durch reife Osteoklasten hemmt, die aus Riesenzelltumoren des Knochens isoliert wurden. Es wird angenommen, dass die extrazelluläre Produktion von lysosomalen Proteasen, die die Knochenresorption vermitteln, durch die Riesenzellen gehemmt wird und die Steroide die Apoptose der osteoklastenähnlichen Zellen auslösen. Infolgedessen hemmt die Verabreichung von Kortikosteroiden den Knochenabbau. Die Tatsache, dass Kortikosteroide bekanntermaßen auch die Knochenresorption fördern und Osteoporose verursachen, steht diesem Erklärungsansatz allerdings entgegen [de Lange et al., 2007].
Zusammenfassung
Auf der Basis aktueller Daten lässt sich vermuten, dass weniger als fünf Prozent aller zentralen Riesenzellgranulome im Rahmen einer RASopathie auftreten [Luna et al., 2022]. Syndromale Läsionen befinden sich bevorzugt im posterioren Unterkiefer, können aber auch bilateral oder sogar multifokal auftreten. In diesem Zusammenhang ist eine Fehldeutung als Cherubismus, als eine andere riesenzellhaltige Läsion sowie als odontogener Tumor oder odontogene Zyste möglich, insbesondere wenn eine zugrundeliegende syndromale Prädisposition nicht in Betracht gezogen wird. In Anbetracht der Seltenheit des syndromalen zentralen Riesenzellgranuloms hängt die Erkennung des zugrundeliegenden Syndroms bei bisher nicht diagnostizierten Personen von der Kenntnis dieser ungewöhnlichen Assoziation und der Identifizierung anderer charakteristischer syndromaler Merkmale ab.
Fazit für die Praxis
Das Noonan-Syndrom ist eines der häufigsten genetischen Syndrome und die zweithäufigste Ursache für angeborene Herzstörungen.
Typische Charakteristika des Noonan-Syndroms sind der Hypertelorismus, die Ptosis sowie große und tiefsitzende Ohren. Die Erkrankung ist häufig mit einem Kleinwuchs und einer Pulmonalstenose vergesellschaftet.
Beim zentralen Riesenzellgranulom handelt es sich um einen gutartigen Knochentumor, der zu Auftreibungen des Kiefers führen kann.
In seltenen Fällen tritt das zentrale Riesenzellgranulom als Symptom des Noonan-Syndroms auf und kann hier sogar bilateral oder multilokulär vorhanden sein.
Die Therapie des zentralen Riesenzellgranuloms besteht meist in der chirurgischen Entfernung. Ist dies nicht möglich, so können alternative Therapieverfahren wie die Gabe von Steroiden, Calcitonin, Interferon oder Denosumab erwogen werden.
Trotz zahlreicher Fallberichte existiert zum aktuellen Zeitpunkt keine nicht-chirurgische Therapie, die bei allen Patienten gleichermaßen wirkungsvoll ist. Dies beruht vermutlich auf der Problematik, dass keines der therapeutischen Regime einen direkten Effekt auf die proliferierenden stromalen Zellen besitzt, sondern vielmehr die osteoklastenähnlichen Riesenzellen beeinflusst. Insbesondere für ein Ansprechen auf die Therapie mit Steroiden scheint die prätherapeutische Analyse auf das Vorliegen eines entsprechend Rezeptors vielversprechend zu sein [de Lange et al., 2007].
Literaturliste
Bufalino A, M Carrera, R Carlos and RD Coletta: Giant cell lesions in noonan syndrome: case report and review of the literature. Head Neck Pathol 4:174-177, 2010.
de Lange J, HP van den Akker and H van den Berg: Central giant cell granuloma of the jaw: a review of the literature with emphasis on therapy options. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 104:603-615, 2007.
EI-Naggar AK, JKC Chan, JR Grandis, T Takata and PJ Slootweg (2017). WHO Classification of Head and Neck Tumors Lyon, International Agency for Research on Cancer (IARC).
Gundlach KKH (2022). Periphere und zentrale sogenannte Kiefergranulome. Mund-Kiefer-Gesichtschirurgie. H.-H. Horch and A. Neff. München, Urban & Fischer in Elsevier 5: 307–318.
Hebron KE, ER Hernandez and ME Yohe: The RASopathies: from pathogenetics to therapeutics. Dis Model Mech 15:2022.
Jacoway JR, FV Howell and BC Terry: Central giant cell granuloma: an alternative to surgical therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 66:572, 1988.
Kämmerer P and M Kunkel, . Peripheres Riesenzellgranulom mit zentraler Kalzifikation. Zahnärztliche Mitteilungen 98:40-42, 2008.
Luna M, N Wolsefer, CX Zambrano and IJ Stojanov: Giant Cell Lesions of the Jaws Involving RASopathy Syndromes. Acta Stomatol Croat 56:77-88, 2022.
Roberts AE, JE Allanson, M Tartaglia and BD Gelb: Noonan syndrome. Lancet 381:333-342, 2013.
Schwarting M, CL Hell and PW Kämmerer: Der besondere Fall mit CME: Peripheres Riesenzellgranulom im Oberkiefer. Zahnärztliche Mitteilungen 110:36-39, 2020.
Terry B and J Jacoway: Management of central giant cell lesions: an alternative to surgical therapy. Oral Maxillofac Surg Clin N Am 6:579–600, 1994.
van der Burgt I: Noonan syndrome. Orphanet J Rare Dis 2:4, 2007.
Zenker M, T Edouard, JC Blair and M Cappa: Noonan syndrome: improving recognition and diagnosis. Arch Dis Child 107:1073-1078, 2022.
CME Info
Titel der Fortbildung: Raumforderung des Kiefers führt zur Erstdiagnose des Noonan-Syndroms
Verfügbar bis: 15.02.24
Bitte loggen Sie sich ein, um weitere Details zu sehen

Dr. med. Dr. med. dent. Diana Heimes
und Gesichtschirurgie – Plastische
Operationen, Universitätsmedizin Mainz
Augustusplatz 2, 55131 Mainz
Dr. med. Francesca Alt
medizin, Schwerpunktbezeichnung
Kinder-Hämatologie und Onkologie,
Palliativmedizin,
Klinik und Poliklinik für Kinder-
und Jugendmedizin
Universitätsmedizin Mainz
Langenbeckstraße 1, 55131 Mainz
Dr. med. dent. Manuela Winau
Wilhelmstraße 2a
65719 Hofheim
PD Dr. med. habil. Alexandra Russo
onkologischen Tagesklinik,
Oberärztin der Sektion
Pädiatrische Hämatologie/Onkologie/
Hämostaseologie
Klinik und Poliklinik für Kinder- und
Jugendmedizin
Universitätsmedizin Mainz
Langenbeckstraße 1, 55131 Mainz

Univ.-Prof. Dr. Dr. Peer W. Kämmerer
Stellvertr. Klinikdirektor
Klinik und Poliklinik für Mund-, Kiefer-
und Gesichtschirurgie – Plastische
Operationen, Universitätsmedizin Mainz
Augustusplatz 2, 55131 Mainz


{kind=link}