„Eine Gefahr für die Innovationskraft“
Die europäische Verordnung für Medizinprodukte, die Medical Device Regulation (MDR), muss aus Sicht der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF) dringend in der praktischen Umsetzung verbessert werden.
Laut AWMF führen die aktuellen Regelungen dazu, dass das System von Prüfung und Zulassung überlastet sei. Medizinprodukte könnten aufgrund aufwändiger Re-Zertifizierungsprozesse vom Markt genommen werden, warnt die AWMF. Außerdem befürchtet sie, dass Hürden für die Entwicklung neuer Medizinprodukte die Innovationskraft des Wissenschaftsstandorts Deutschland bedrohen könnten.
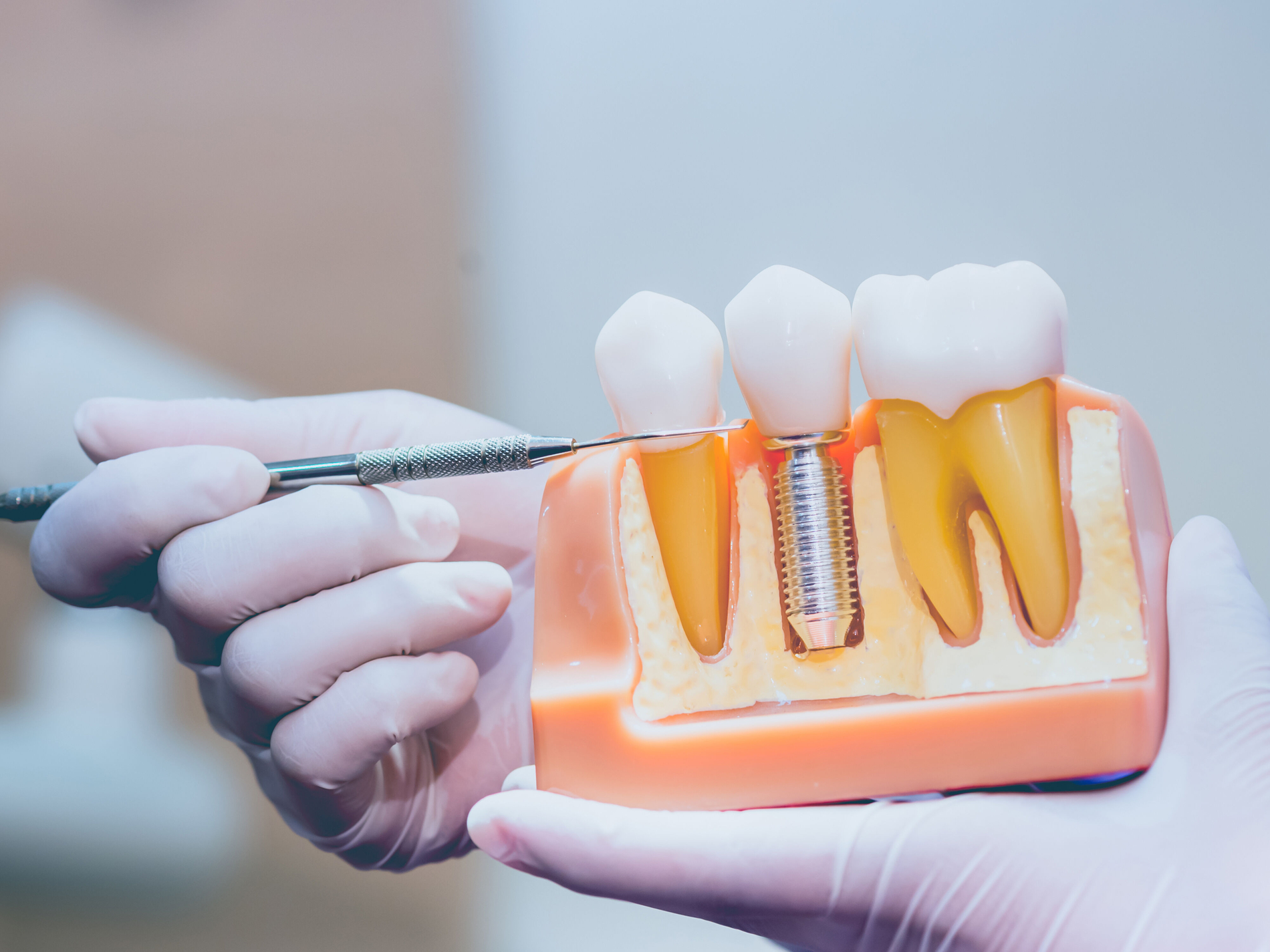
Unnötig: Bereits bewährte Medizinprodukte müssen eine komplette Re-Zertifizierung durchlaufen
Die AWMF fordert deshalb gestaffelte Zulassungsregelungen, Förderprogramme für Registerdaten sowie mehr Investitionen und risikoadaptierte regulatorische Rahmenbedingungen für klinische Studien.
Zwar unterstütze die AWMF das Ziel der Verordnung, die Patientensicherheit durch höhere Anforderungen bei der Marktzulassung von Medizinprodukten wie etwa Herzschrittmachern, Implantaten oder Prothesen zu erhöhen, vorbehaltlos, erklärt Prof. Rolf-Detlef Treede, Präsident der AWMF dazu. Jedoch stellten die derzeitigen Regelungen der MDR große Hürden für die Zulassung von Medizinprodukten dar. Das könne aktuell zu Versorgungslücken und langfristig zur Schwächung der Wissenschaft in Deutschland führen, warnte er und plädierte dafür, die Umsetzung der Verordnung praktikabel zu halten.
Die MDR sieht beispielsweise vor, dass bereits bewährte Medizinprodukte eine komplette Re-Zertifizierung durchlaufen müssen. Zuständig dafür sind sogenannte Benannte Stellen. Die AWMF führt an, dass dies circa 24.000 Produkte betreffe. Das führe zu einer Überlastung der Benannten Stellen sowie zu erheblichem Aufwand für die Medizinproduktehersteller. Diese hätten bereits Produkte vom Markt genommen, von denen einige jedoch für bestimmte Operationen notwendig seien. Hier drohten Versorgungslücken, warnt die AWMF. Um potenzielle Ausfälle von Medizinprodukten mit resultierenden Versorgungslücken aufzudecken und belastbare Daten zu liefern, habe die AWMF ein Melderegister eingeführt.
Häufig fehle es auch an klinischen Daten, die für die Re-Zertifizierung von bereits verfügbaren Medizinprodukten nötig seien, heißt es weiter. Die AWMF schlägt deshalb vor, verstärkt vorliegende Registerdaten für eine niederschwellige Re-Zertifizierung zu nutzen.
Die AWMF sieht in den derzeitigen Vorgaben der Verordnung nicht zuletzt auch die Innovationskraft der Wissenschaft in Deutschland gefährdet. Klinische Studien hätten inzwischen ein erheblich größeres Gewicht für die Zertifizierung von Medizinprodukten gewonnen, argumentiert die AWMF. Gleichzeitig seien auch die Anforderungen an klinische Studien gestiegen. Deshalb fordert sie, die finanziellen Rahmenbedingungen für klinische Studien aus öffentlichen Mitteln zu verbessern, damit die zusätzlichen Aufwände leistbar seien und die Abhängigkeit von Drittmitteln reduziert werde.
Zulassungsprozesse in Europa sind deutlich teurer als in den USA oder Kanada
Eine weitere Gefahr für die Innovationskraft der Wissenschaft am Standort Deutschland besteht aus Sicht der AWMF in den hohen Kosten der Zulassungsprozesse. So sei die Re-Zertifizierung eines auf dem Markt befindlichen Bestandsproduktes in Europa unter MDR-Bedingungen deutlich teurer als in den USA oder Kanada. Auch fehlten den europäischen Medizinprodukteherstellern, bei denen es sich in mehr als 90 Prozent der Fälle um kleine oder mittlere Unternehmen handele, häufig die finanziellen Mittel, um Erstzulassungen durchführen zu lassen. Die AWMF warnte davor, dass Zulassungsstudien für innovative Produkte zukünftig primär außerhalb Europas durchgeführt werden könnten. Die Folge sei, dass diese Produkte erst spät für die Krankenversorgung in Europa zur Verfügung stehen könnten. Regularien für Zulassungsstudien müssten auf ein leistbares Maß reduziert werden, forderte sie.
Die Rezertifizierung von Medizinprodukten soll in der EU eigentlich bis Mai 2024 abgeschlossen sein. Vor kurzem wurde bekannt, dass die zypriotische EU-Gesundheitskommissarin Stella Kyriakides eine Verschiebung der Frist in Aussicht gestellt und einen entsprechenden Legislativvorschlag für den Januar 2023 angekündigt hat.
")
