Fibröse Dysplasie des Os temporale und der Kiefergelenksregion
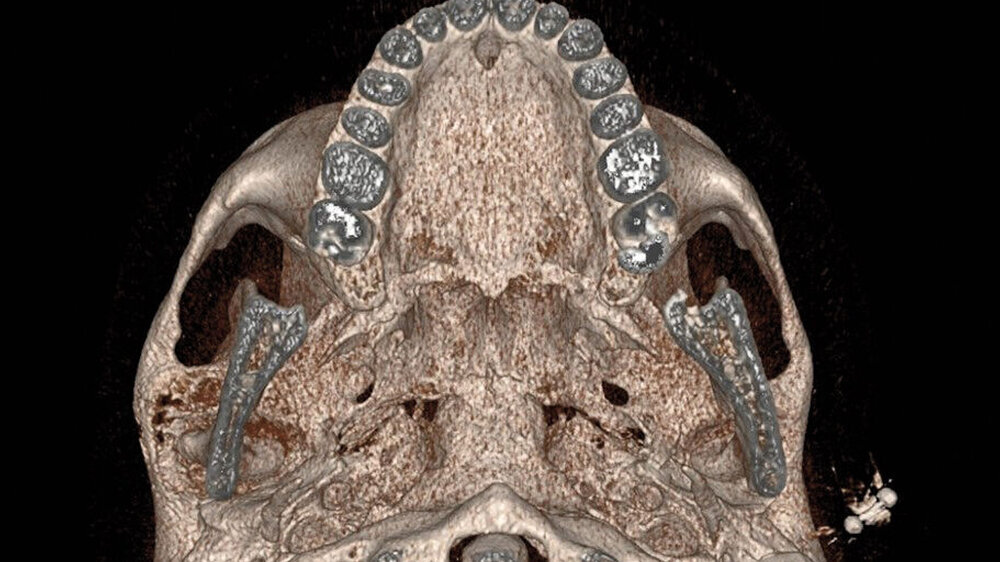

Eine 34-jährige Patientin wurde im Rahmen einer Routineuntersuchung ohne Beschwerden auf Anraten ihrer behandelnden Zahnärztin vorgestellt. Zufällig wurde bei bildgebenden Verfahren eine Läsion im Bereich des Os temporale und der Kiefergelenksregion rechts entdeckt.
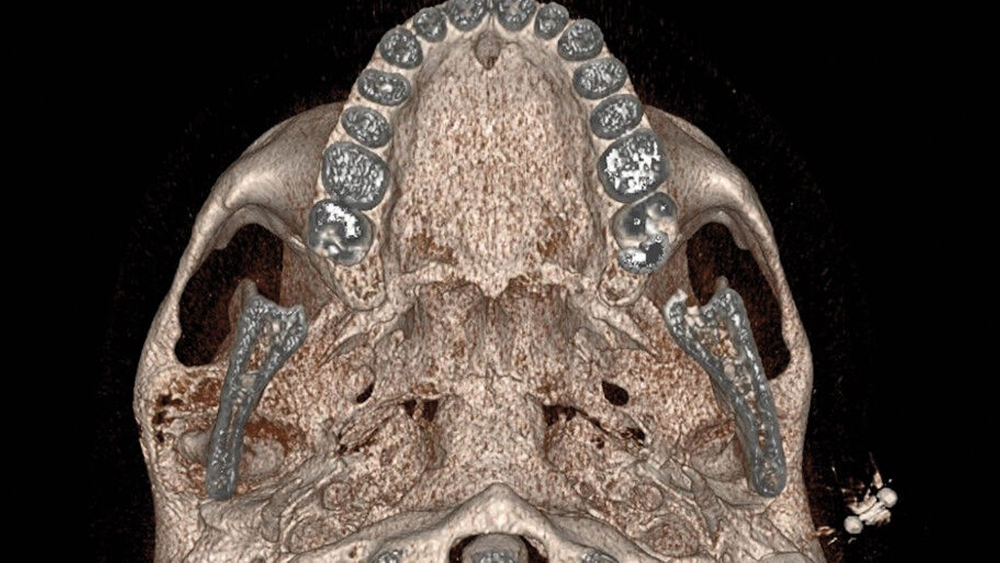
Im MRT und CT zeigte sich eine expansiv wachsende, überwiegend mattglasartige Knochenveränderung mit osteolytischen Anteilen und Unterbrechungen der Knochenstruktur, insbesondere in den unteren Partien nahe dem Kiefergelenk. Die Läsion war mit vermehrter intraossärer Kontrastmittelanreicherung assoziiert, was für eine fibröse Dysplasie spricht. Das Kieferköpfchen war abgeflacht, und es lag eine geringe Flüssigkeitseinlagerung im Kiefergelenk vor. In der Bildgebung fanden sich keine Hinweise auf eine Beeinträchtigung der umliegenden Foramina oder angrenzender Knochenstrukturen wie des Keilbeins (Abbildung 1).
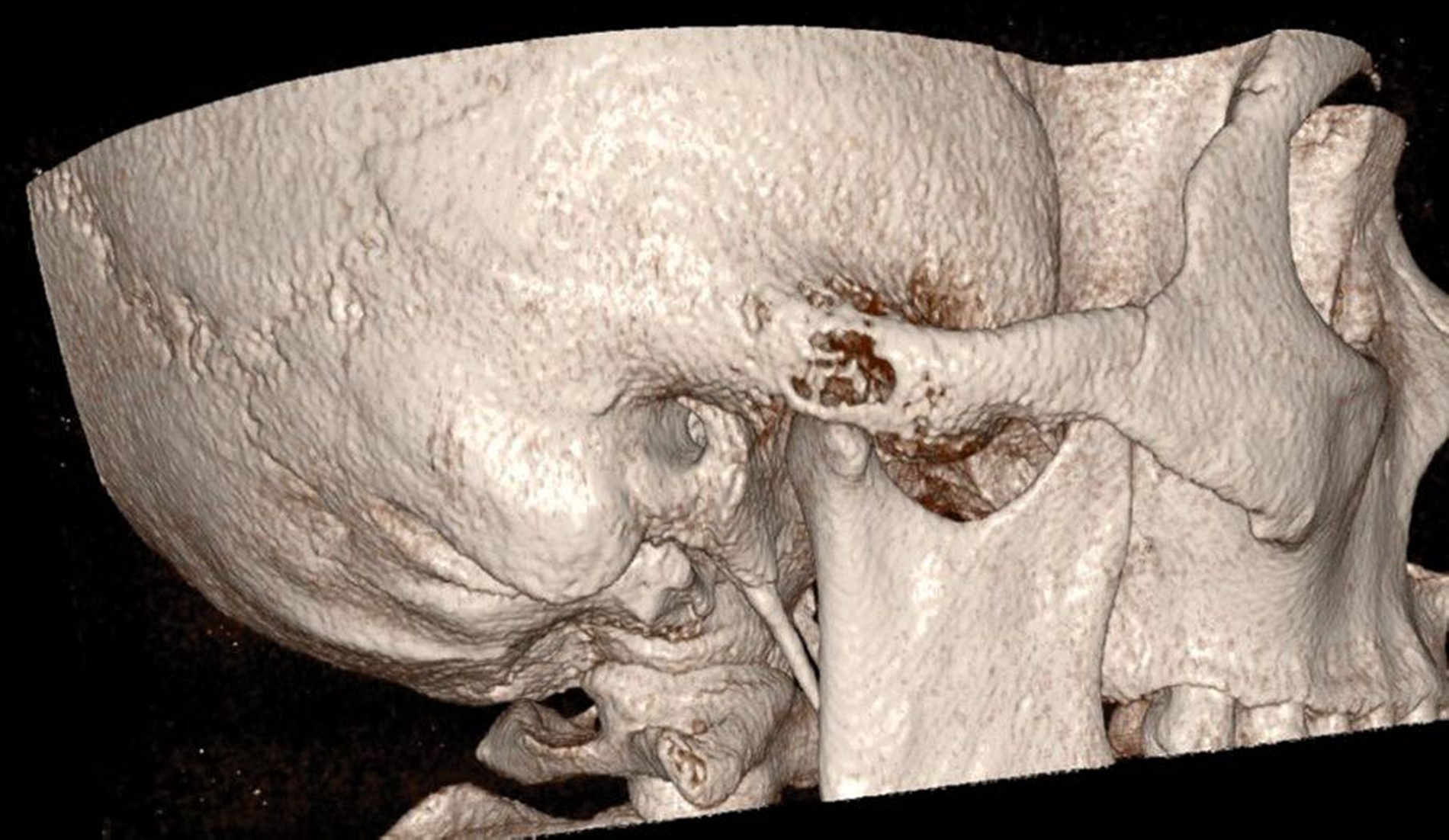
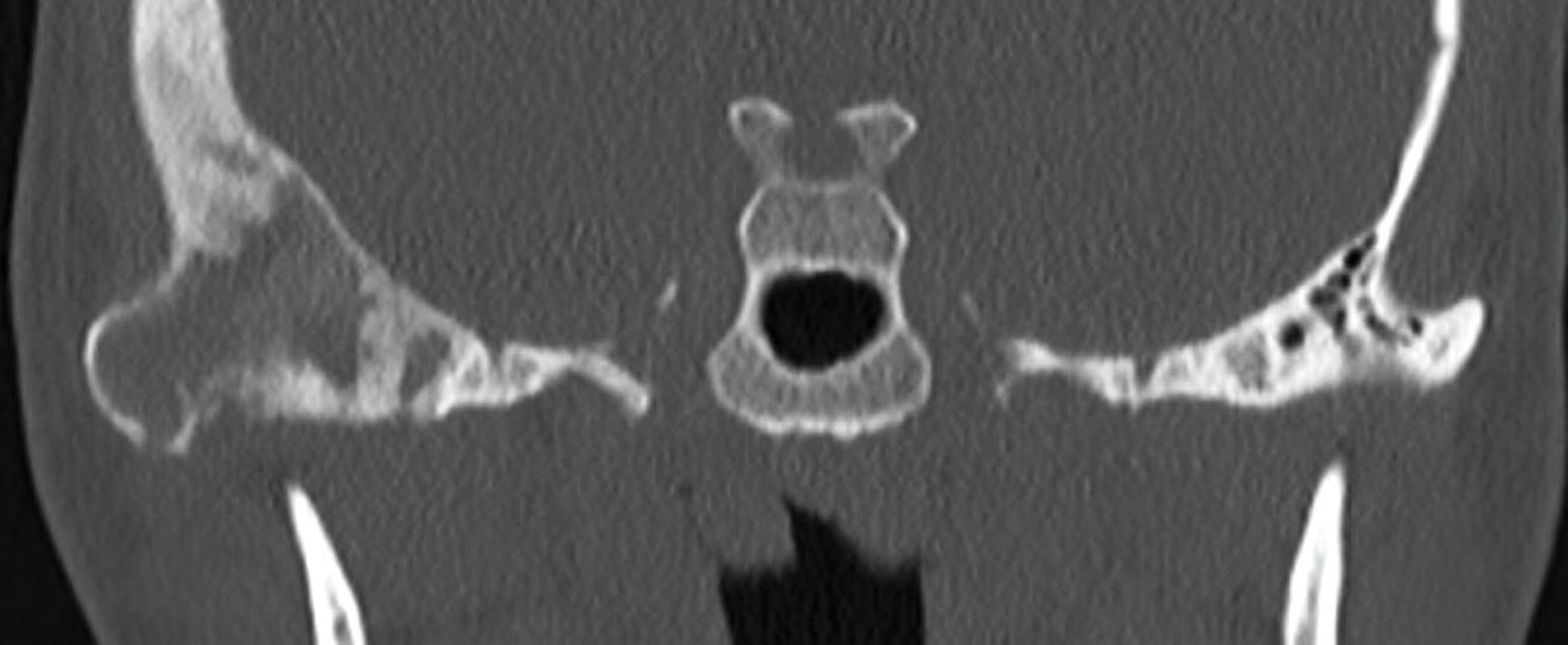
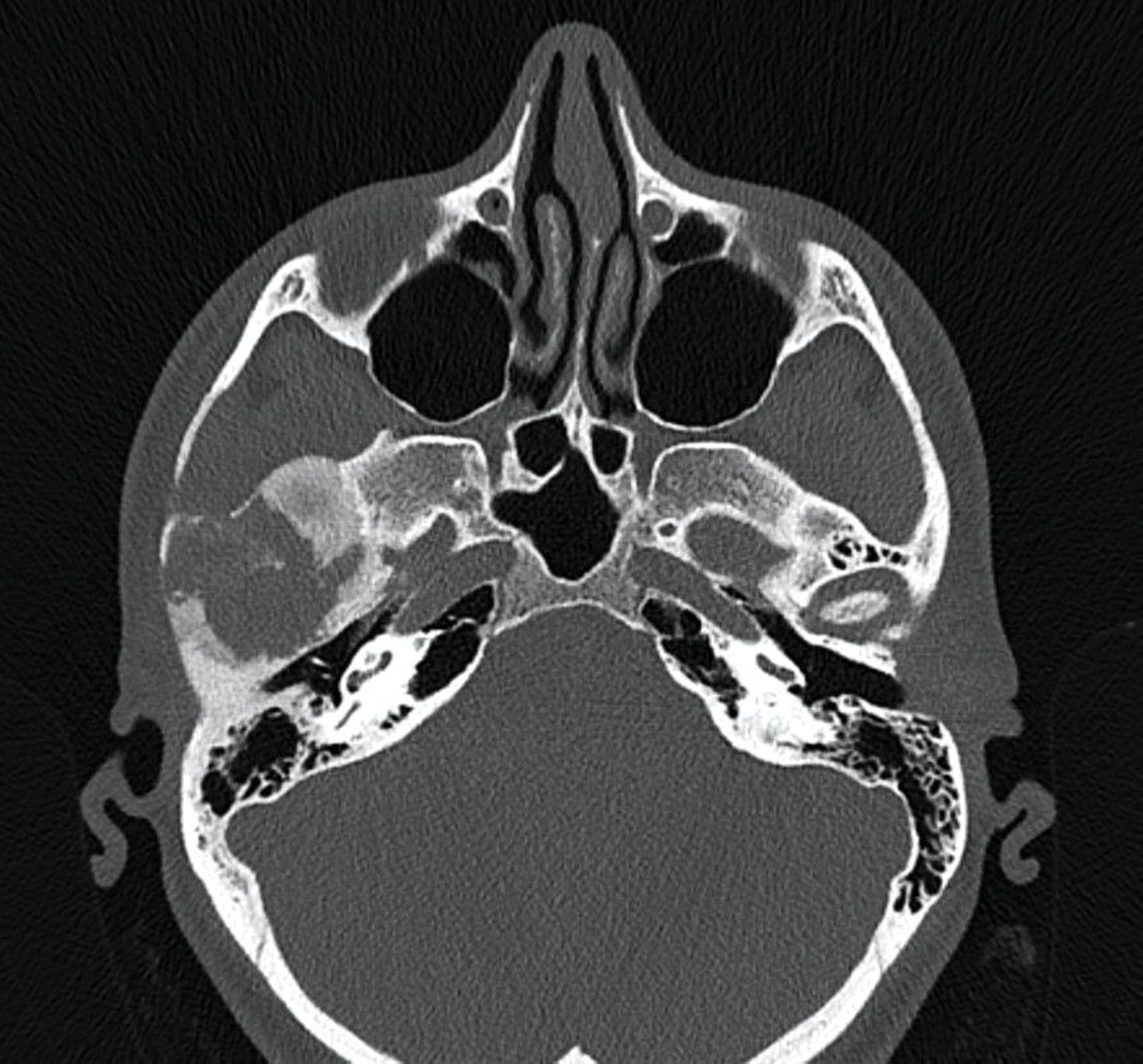
Der Vergleich mit früheren Bildgebungen (MRT 2019 und ältere Schädelröntgenaufnahmen) zeigte ein leicht progredientes Wachstum der Läsion über mehrere Jahre hinweg. Die bildgebenden Befunde und hier insbesondere das umfangreiche Ausmaß der Osteolysen führten zur Empfehlung einer gezielten Biopsie, um eine Malignitätsdiagnose (zum Beispiel Osteosarkom oder Chondrosarkom; aber auch maligne Entartung einer fibrösen Dysplasie) auszuschließen und die Läsion histologisch zu charakterisieren.
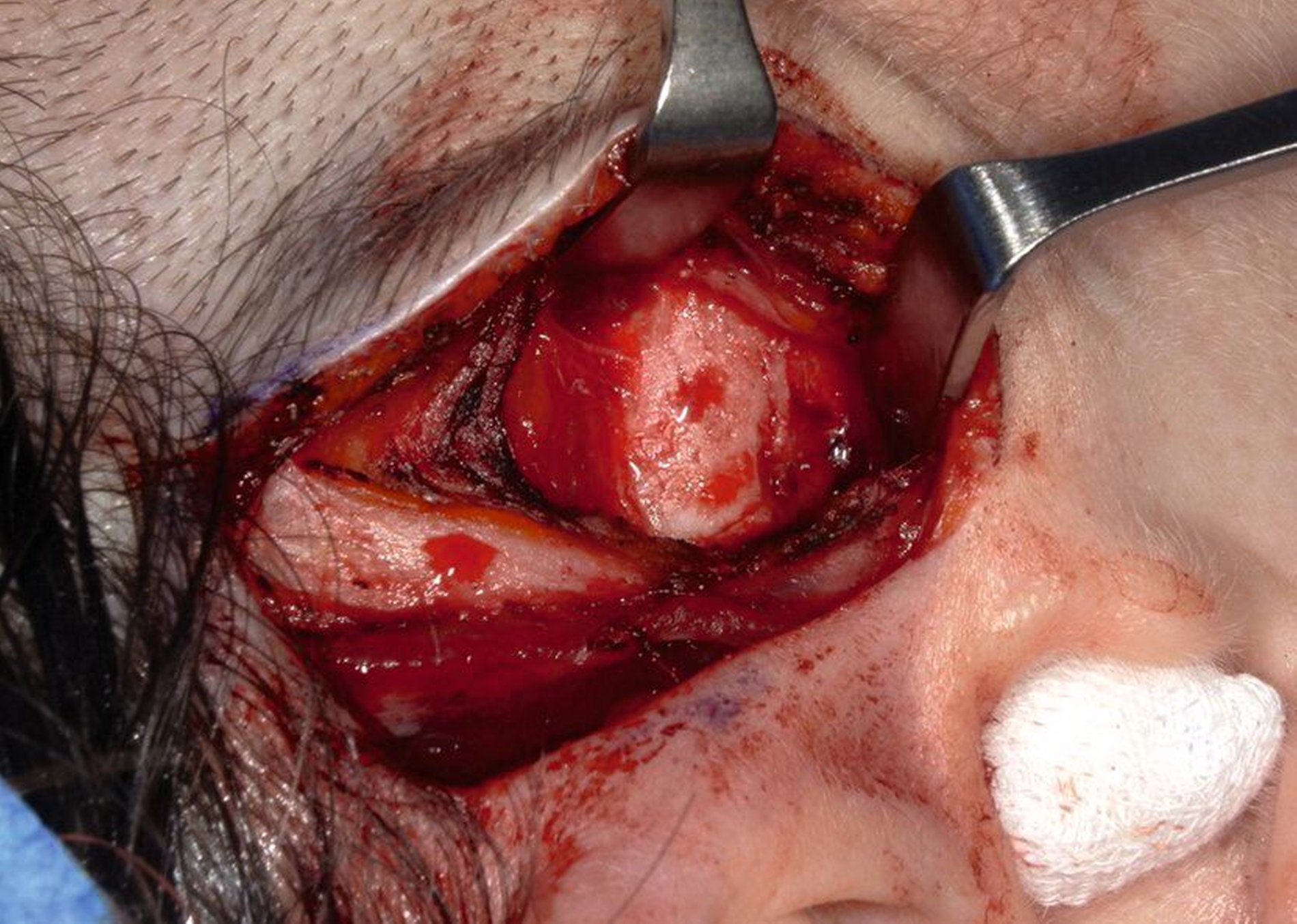
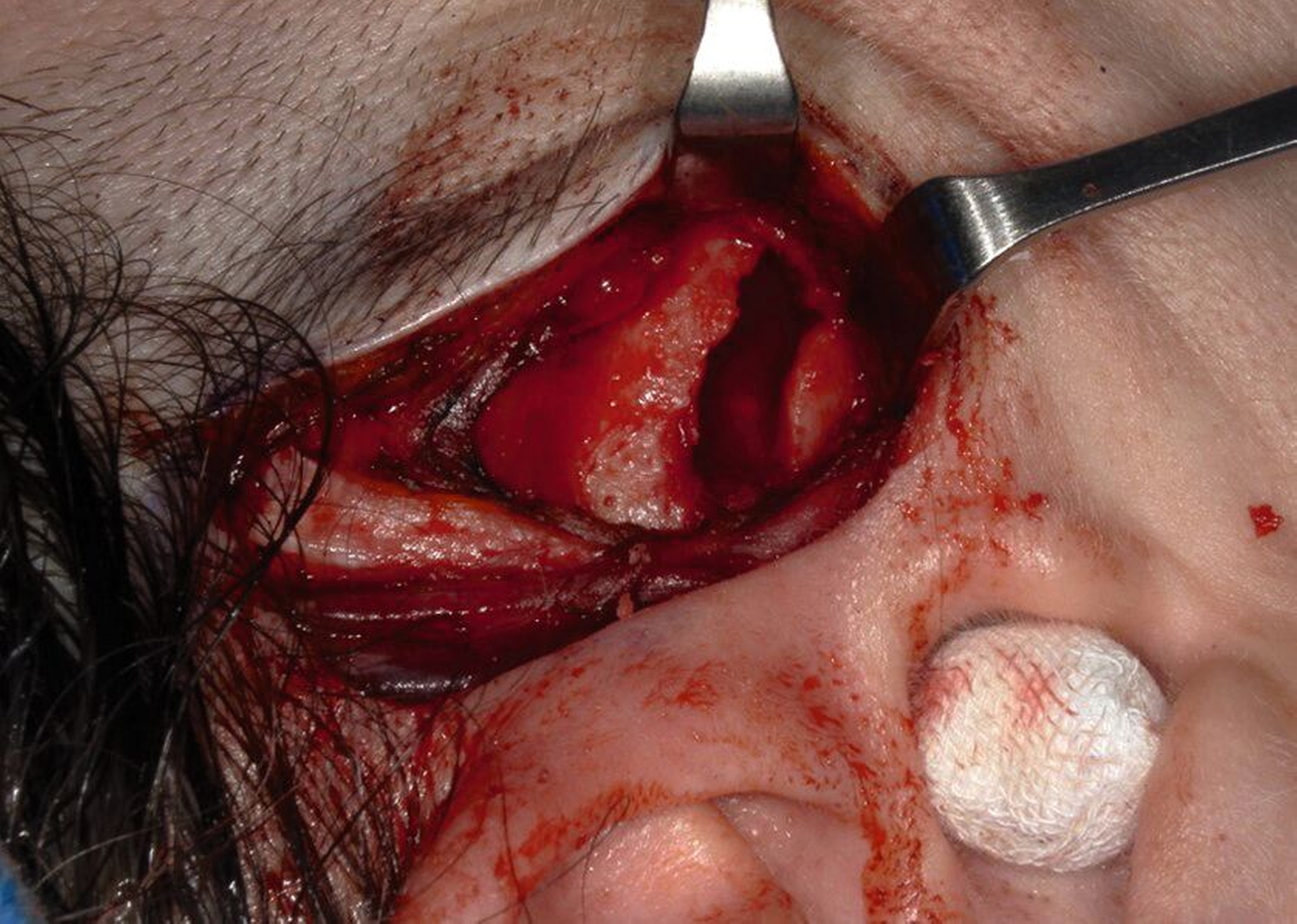
Der operative Eingriff erfolgte in Intubationsnarkose über einen präaurikulären Zugang im kranialen Schläfenbogen (Abbildung 2). Nach stumpfer Präparation und Darstellung der Fossa mandibularis zeigte sich intraoperativ ein kortikal durchbrochenes, strukturell verändertes Knochenareal mit typischer Konsistenz einer fibrösen Dysplasie (Abbildung 3). Es wurde eine gezielte knöcherne Biopsie entnommen (Abbildung 4), wobei keine Infiltration der angrenzenden Weichteilstrukturen oder kritischer anatomischer Nachbarbereiche erkennbar war. Der Eingriff verlief komplikationslos. Die intraoperative Blutung war gering, und der Defekt wurde mit Kollagenschwämmen augmentiert. Der Wundverschluss erfolgte schichtweise. Die postoperative Heilung war regelrecht.
Histologisch fand sich fibrokollagenäres Gewebe mit irregulär-dysplastischen Knochentrabekeln ohne Atypien oder Nekrosen in einem fibrösen Stroma. Molekulargenetisch wurde mittels Next Generation Sequencing eine pathogene GNAS-Mutation (p.R844H) nachgewiesen, die typisch für eine fibröse Dysplasie ist. Die Diagnose einer fibrösen Dysplasie des Os temporale und der Kiefergelenksregion wurde auch unter Einbezug der Referenzpathologie in Basel bestätigt. Aufgrund der asymptomatischen Klinik und der stabilen bis langsam progredienten Läsion wurde von einer aggressiven Therapie abgesehen, und die Patientin wird konservativ überwacht.
Diskussion
Die fibröse Dysplasie (FD) ist eine seltene (1:4.000 bis 1:10.000), benigne, nicht-erbliche fibro-ossäre Läsion des Knochens, die durch eine abnorme Knochen- und Bindegewebsbildung charakterisiert ist. Das Diagnosealter variiert aufgrund unterschiedlicher klinischer Präsentationen, liegt jedoch typischerweise innerhalb der ersten vier Lebensjahrzehnte [Soluk-Tekkesin et al., 2022; Tuompo et al., 2025]. Verursacht wird die FD durch eine somatische postzygotische Mutation im GNAS-Gen [Tuompo et al., 2025].
Diese Mutation, typischerweise an der Position R201 (R201H oder R201C), führt zu einer konstitutiven Aktivierung der Gs-Protein-vermittelten Signaltransduktion und einer gesteigerten intrazellulären cAMP-Produktion [Golden et al., 2025]. In der Folge kommt es zu einer gestörten Reifung der Osteoblasten und einem Ersatz des normalen Lamellenknochens durch unreifes, fibroossäres Gewebe mit ungeordneten Knochenbälkchen in fibrösem Stroma. Die Mutation entsteht früh embryonal, wodurch das klinische Bild durch den Zeitpunkt der Mutation und das betroffene Keimblatt determiniert wird [Xue et al., 2025].
Klinisch wird zwischen einer monostotischen Form (circa 70 bis 85 Prozent der Fälle) und einer polyostotischen Ausprägung (15 bis 30 Prozent) unterschieden. Erstere ist oft ein Zufallsbefund bei jungen Erwachsenen, letztere manifestiert sich meist bereits im Kindesalter und kann mit hormonellen Störungen einhergehen, wie sie beim McCune-Albright-Syndrom auftreten. Dieses Syndrom umfasst eine triadische Kombination aus polyostotischer FD, kutanen Café-au-lait-Flecken und endokrinologischen Dysfunktionen, insbesondere einer Pubertas praecox, seltener Hyperthyreose oder Wachstumshormonüberschuss [Priego Zurita et al., 2025].
Die FD betrifft überwiegend die Schädel- und Gesichtsknochen. Kraniofaziale Läsionen finden sich bei etwa zehn bis 25 Prozent der Patientinnen und Patienten mit monostotischer FD und bei bis zu 50 Prozent der Patienten mit polyostotischer FD [Cheng et al., 2012]. Die klinische Präsentation der kraniofazialen FD (CFD) variiert erheblich in Abhängigkeit von der Lokalisation und der Ausdehnung der Läsionen. Die Symptome entstehen in der Regel durch strukturelle Deformierungen und den Druck auf angrenzende Gewebe. Die zygomatico-maxilläre Region scheint am häufigsten betroffen zu sein, während Läsionen im Bereich des Os temporale und der Kiefergelenksregion – wie in diesem Fall – nur selten beschrieben werden. Die typischen Symptome umfassen schmerzlose Knochenschwellungen, eine faziale Asymmetrie und Kopfschmerzen sowie sensible Ausfälle infolge kompressiver Neuropathien. In seltenen Fällen kommt es zur Kompression vitaler intrakranieller Strukturen – mit schwerwiegenden Folgen [Jaulent et al., 2019]. Dennoch bleiben die meisten Patientinnen und Patienten mit CFD asymptomatisch, und die Diagnose wird – wie auch im hier vorgestellten Fall – häufig zufällig gestellt [Tuompo et al., 2025].
Monostotische CFD-Läsionen, das heißt eine Erkrankung, die ausschließlich das kraniofaziale Skelett betrifft, wachsen in der Regel proportional zum Skelettwachstum und kommen zum Stillstand, sobald die skelettale Reife erreicht ist. Frühere Arbeiten berichten jedoch, dass sich die Läsionen in einigen Fällen auch im Erwachsenenalter weiterentwickeln, vergrößern und aktiv bleiben können – was zu fortschreitenden Deformitäten, häufigeren Frakturen und einer höheren Krankheitslast führt [Lee et al., 2012]. Dies tritt häufiger bei der polyostotischen FD auf, die mit einer stärkeren skelettalen Beteiligung und diffuseren Läsionen an mehreren Skelettabschnitten einhergeht. Mitunter können Läsionen auch nach einer langen inaktiven Phase reaktiviert werden, etwa durch hormonelle Veränderungen während der Schwangerschaft. Fallberichte beschreiben zudem eine Reaktivierung kraniofazialer Läsionen nach Traumata oder operativen Eingriffen [Pardo-Maza et al., 2015; Tuompo et al., 2025].
Die Diagnose der CFD basiert in der Regel auf bildgebenden Verfahren, insbesondere der Computertomografie. Typisch ist dabei das sogenannte milchglasartige Erscheinungsbild des geflechtartigen Knochens, wie im vorgestellten Fall. Allerdings können die Läsionen sehr unterschiedlich erscheinen und auch sklerotische, zystische oder gemischte Strukturen aufweisen. Zu den differenzialdiagnostischen Überlegungen zählen unter anderem Meningeome, die Paget-Krankheit, niedriggradige Osteosarkome sowie verschiedene benigne fibroossäre Läsionen. Bei CFD-Läsionen mit maligner Entartung werden in der Bildgebung eine Destruktion der Kortikalis und eine Zunahme der Ausdehnung ins Weichgewebe beschrieben [Sun et al., 2014].
Die maligne Transformation bei FD ist jedoch insgesamt äußerst selten und wird in der Literatur mit einer Häufigkeit von etwa 0,4 bis vier Prozent angegeben – bei den meisten Studien liegt der Wert deutlich unter ein Prozent, insbesondere bei der monostotischen Form, während polyostotische Erscheinungsformen und das McCune‑Albright‑Syndrom ein etwas erhöhtes Risiko aufweisen [Müller et al., 2024; Tuompo et al., 2025]. Wenn klinische Befunde und bildgebende Ergebnisse keine eindeutige Diagnose ermöglichen, kann eine histologische Untersuchung zur Bestätigung herangezogen werden. Dabei zeigen sich typischerweise dysplastische Knochentrabekel in einem fibrösen Stroma.
Das Management der CFD richtet sich nach dem individuellen Patientenprofil, der Lokalisation und der Ausdehnung der Läsion sowie den damit verbundenen Symptomen. Da das Wachstum der Läsionen in der Regel langsam verläuft und die Mehrheit der Patienten asymptomatisch ist, erfolgt die Behandlung häufig konservativ in Form einer regelmäßigen Beobachtung. Bei symptomatischen, aktiven oder progredienten Läsionen kann hingegen ein proaktives Vorgehen mit einem individuell abgestimmten, interdisziplinären Therapieansatz erforderlich sein, der sich an den betroffenen kraniofazialen Strukturen orientiert.
Das Nachsorgeintervall und die Art der Nachsorge richten sich nach dem Verhalten und der Ausdehnung der Erkrankung. Sie kann serielle bildgebende Untersuchungen, Tests des Hör- und Sehvermögens oder anderer funktioneller Parameter sowie zahnärztliche Kontrollen umfassen. Eine Veränderung des Läsionsverhaltens, insbesondere ein neu auftretendes aggressives Wachstum, sollte den Verdacht auf eine maligne Transformation wecken [Pack et al., 2016; Tuompo et al., 2025].
Fazit für die Praxis
Die kraniofaziale fibröse Dysplasie (CFD) ist selten, verläuft oft asymptomatisch und wird häufig zufällig entdeckt, auch in der zahnärztlichen Bildgebung.
Typisches Leitsymptom ist eine schmerzlose, langsam progrediente knöcherne Raumforderung, insbesondere im Bereich der Maxilla oder des Os zygomaticum.
Die CT mit typischem milchglasartigem Knochenmuster ist die zentrale diagnostische Maßnahme.
Asymptomatische Patientinnen und Patienten werden in der Regel konservativ betreut und der Krankheitsverlauf ist meist stabil. Dennoch sind regelmäßige zahnärztliche und bildgebende Kontrollen wichtig, da es auch Jahre nach der Erstmanifestation zu einer Reaktivierung kommen kann.
Ein interdisziplinärer Therapieansatz ist essenziell, insbesondere bei ästhetisch oder funktionell relevanten Läsionen. Zahnärztinnen und Zahnärzte spielen eine zentrale Rolle in der Verlaufskontrolle, der Erkennung dentaler oder funktioneller Komplikationen und der rechtzeitigen Einbindung spezialisierter Fachdisziplinen.
Intravenös verabreichte Bisphosphonate stellen die medikamentöse Standardtherapie zur Schmerzbehandlung sowie zur Vermeidung von Läsionswachstum, Frakturen und einem postoperativen Rezidiv dar und können prinzipiell bei allen Formen der FD eingesetzt werden. Die klinischen Ergebnisse sind jedoch uneinheitlich [Couturier et al., 2017]. Chirurgische Eingriffe dienen der Wiederherstellung von Funktion, der Verringerung von Deformitäten, der Behandlung von durch die Läsion verursachten Schmerzen und geschehen insbesondere im kraniofazialen Bereich aus ästhetischen Gründen [Zeng et al., 2013]. Eine radikale Resektion gilt bei monostotischer Erkrankung als einzige kurative Therapieoption. In anatomisch komplexen Regionen kann hingegen ein konturierendes Abtragen („Shaving“) und Remodellieren der Läsion vorgenommen werden, da diese Verfahren in der Regel schneller, kosteneffizienter und mit einer kürzeren Erholungszeit verbunden sind. Eine nicht-radikale Resektion birgt jedoch das Risiko eines Rezidivwachstums. insbesondere bei polyostotischer FD oder beim McCune-Albright-Syndrom sowie bei Eingriffen während des Skelettwachstums [Park et al., 2020].
Literaturliste
Cheng J, Y Wang, H Yu, D Wang, J Ye, H Jiang, Y Wu and G Shen: An epidemiological and clinical analysis of craniomaxillofacial fibrous dysplasia in a Chinese population. Orphanet J Rare Dis 7:80, 2012.
Couturier A, O Aumaitre, L Gilain, B Jean, T Mom and M Andre: Craniofacial fibrous dysplasia: A 10-case series. Eur Ann Otorhinolaryngol Head Neck Dis 134:229-235, 2017.
Golden BM, SK Tucker, M Carpenter, M Santi, AN Viaene, WH Peranteau, JW Swanson, SP Bartlett, JA Taylor and EC Liao: Integration of Clinical, Radiographic, Histologic, and Molecular Findings to Diagnose Craniofacial Fibrous Dysplasia. J Craniofac Surg 2025.
Jaulent P, E Vignot and R Chapurlat: Fibrous dysplasia of occipital bone revealed by acute intracranial hypertension. Osteoporos Int 30:691-693, 2019.
Lee JS, EJ FitzGibbon, YR Chen, HJ Kim, LR Lustig, SO Akintoye, MT Collins and LB Kaban: Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis 7 Suppl 1:S2, 2012.
Müller D, N Aljinovic Ratkovic, I Blivajs, D Franceski, K Krstanac and S Seiwerth: Osteosarcoma in a Patient with Cranial Fibrous Dysplasia: A Case Report. Acta Clin Croat 63:191-196, 2024.
Pack SE, AA Al Share, FA Quereshy and DA Baur: Osteosarcoma of the Mandible Arising in Fibrous Dysplasia-A Case Report. J Oral Maxillofac Surg 74:2229 e2221-2229 e2224, 2016.
Pardo-Maza A, L Lassaletta, E Ruiz-Bravo, R Perez-Mora, J Penarrocha and J Gavilan: Fibrous dysplasia of the temporal bone secondary to ear surgery: a case report. J Med Case Rep 9:129, 2015.
Park JW, JH Jung, SJ Park and SY Lim: Evaluation of natural growth rate and recommended age for shaving procedure by volumetric analysis of craniofacial fibrous dysplasia. Head Neck 42:2863-2871, 2020.
Priego Zurita AL, OO Bulaicon, J Bryce, N Arrieta, M Caballero Campos, M Cherenko, G Doxiadis, C Grasemann, MK Javaid, H McDevitt, SW van der Meeren, D Ovejero Crespo, L de Sanctis, L Seefried, AA Verrijn Stuart, D Tessaris, PB de Witte, R Chapurlat, SF Ahmed and NM Appelman-Dijkstra: Developing a Standardised Dataset for Natural History Studies in Fibrous Dysplasia/McCune-Albright Syndrome. Calcif Tissue Int 116:68, 2025.
Soluk-Tekkesin M, A Sinanoglu, F Selvi, H Cakir Karabas and N Aksakalli: The importance of clinical and radiological findings for the definitive histopathologic diagnosis of benign fibro-osseous lesions of the jaws: Study of 276 cases. J Stomatol Oral Maxillofac Surg 123:364-371, 2022.
Sun TT, XF Tao and HM Shi: Spontaneous osteosarcoma in craniomaxillofacial fibrous dysplasia: clinical and computed tomographic features in 8 cases. Oral Surg Oral Med Oral Pathol Oral Radiol 118:e24-31, 2014.
Tuompo S, RE Makitie and MT Nieminen: Craniofacial fibrous dysplasia: A review of current literature. Bone 192:117377, 2025.
Xue J, J Zhang, M Ma, X Li, L Sun, R Shi and T Li: Identification of Novel and Rare GNAS Mutations in Craniofacial Fibrous Dysplasia. J Oral Pathol Med 54:120-125, 2025.
Zeng HF, JJ Lu, L Teng, XL Jin, JJ Xu, C Zhang, MB Xu, F Xie, T Tian, R Xu and HH Wu: Surgical treatment of craniomaxillofacial fibrous dysplasia: functionally or aesthetically? J Craniofac Surg 24:758-762, 2013.
CME Info
Titel der Fortbildung: Fibröse Dysplasie des Os temporale und der Kiefergelenksregion
Verfügbar bis: 31.12.25
Bitte loggen Sie sich ein, um weitere Details zu sehen

Univ.-Prof. Dr. Dr. Peer W. Kämmerer
Stellvertr. Klinikdirektor
Klinik und Poliklinik für Mund-, Kiefer-
und Gesichtschirurgie – Plastische
Operationen, Universitätsmedizin Mainz
Augustusplatz 2, 55131 Mainz


{kind=link}